Pouring LB plates from prepped media-PDF
Notes
- Melt LB ahead of time because it cools to 55 degrees slowly. A good way to do this is melt it in the morning then leave it in the 60 degree incubator or a 60 degree bath. If in a rush, do not attempt to cool the media rapidly by running cold water over the container as this can cause the glass to shatter. Instead, media can be left at room temperature to cool more rapidly.
Materials
- LB Agar 1.2% (from the media room)
- Empty plates (above any of the -20 freezers)
- antibiotics (liquid stocks from our -20 freezer)
Method
Preparation of LB Agar
- Obtain 1.2% LB agar from the media room (500 ml per bottle).
- Be sure to sign it out
- Melt in microwave:
- loosen the cap
- use 50% power (enter time, press Power, 5, Start)
- monitor as you melt
- takes approx. 10 minutes per bottle.
- If too hot, let the melted solution cool so that it’s warm, but not hot, to the touch – you can use the water bath to do this (50-60 degrees); this won’t let it resolidify. At room temp. it may take 20 mins for LB to cool sufficiently.
- Once it’s cool enough (50-60 degrees) – add antibiotics: final concentration, 50ug/ml AMP; 20ug/ml KAN. (concentrations of liquid stocks: Amp; 50mg/ml. Kan, 10mg/ml.) For 500mL of LB Agar use:
- 500μL Amp stock
- 1mL Kan stock
- Swirl to mix; try not to make many bubbles.
- I usually let sit @ 55C for 3-4 min after swirling to let the bubbles go down.
Actual Pouring
- Obtain a container of empty plates. One bottle (500ml) of LB Agar will make about one container of plates (20 plates).
- Using sterile technique (flame the top of the bottle)(why? what are you killing here? the plastic sealing ring?), pour the LB Agar into the plates.
- Cover the base of the plate, and then just a bit more after that.
- Recap each plate upon pouring. If there are lots of bubbles in your plates (i.e., more than one or two on the edge), you can flame the plate using the small bunsen burner to eliminate bubbles. (See a demo on this). Another way to remove the fine bubbles that may be in your flask before pouring is to mist the inside of the flask with a 75% ethanol spray bottle.
- Leave plates to dry and cool for a while (overnight even).
- It is a good idea to label the stack of plates to indicate antibiotic.
- Store the plates in their original bags – upside down, so that the gel is hanging downwards (this keeps condensation off the gel).
- Label the bags following taping rules:
- Red tape in back (indicates LB)
- Tape in front indicates antibiotic (green=Kan, yellow=Amp, more taping/color rules are on the refrigerator in 68-564)
- Also write name of antibiotic, and concentration, on the front piece of tape.
- Store the labeled bags of plates in the cold room.
Nested RT-PCR-PDF

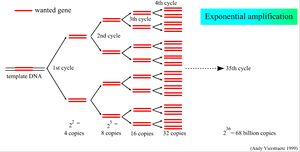 Polymerase dependent expansion of a DNA amplicon
Polymerase dependent expansion of a DNA amplicon
- Korey Griffin Resource List
qPCR Advantages
- Collect reliable data at low cycle numbers. Traditional PCR is less sensitive/ must proceed to higher Ct observe on EtBr agarose gel.
- qPCR (real time) data are captured the duration of low cycle number and minimizes error.
- Earlier and more sensitive early cycle measurements in the amplification process mitigate substrate exhaustion (dNTPs).
Ct (threshold cycle) value
Ct (threshold cycle) value is cycle number at which fluorescence emission (SYBR Green) emerges from the fluorescence threshold. Fluorescence threshold signal represents an emission (enrichment) output above background fluorescence. At Ct (threshold cycle), amplicon product at a detectable amount is entering the early exponential phase of expansion.
- The lower a Ct value, the higher amount of starting substrate (parent template).
- Ct (threshold cycle) are inversely proportional to basal expression of target nucleic acid (mRNA)
- Ct (threshold cycle) minimum fluorescence detection sensitivity is reaents, primers, and qPCR apparatus dependent.
- The cycle number at threshold value detection =cycle threshold (Ct).
Ct <25 = positive/abundant mRNA Ct 25-30 = positive/moderate mRNA Ct 30-40 = negative/minimal mRNA/false positive/contamination
SYBR Green
- SYBR Green alone on fluorescent plate reader at 497 nanometer blue light excitation fluoresces =background fluorescence.
- SYBR Green intercalates into dsDNA, and with robust fluorescence @(λmax = 497 nm) emits @ band pass (λmax = 520 nm).
- One unit of dsDNA to SYBR Green = one unit of fluorescence (after subtracting background fluorescence in excess amount of SYBR Green substrate). Two units of DNA = 2x as much fluorescence.
Traditional (Nested) PCR overview
Santa Cruz Biotechnology inc. offers nested primers for measuring transcript levels. Each product will have 2 vials, an A set (forward & reverse) and B set (forward & reverse) primers. Each set of primers is provided 20 µl at a concentration of 10 µM. Since the primers are designed toward an mRNA template, they are designed to cross exon junctions and will not amplify genomic DNA.
The amplification of RNA requires the conversion of the RNA substrate into DNA. This is achieved through the use of a reverse transcriptase such as AMV RT (avian myeloblastis virus reverse transcriptase) or M-MuLV RT (moloney murine leukemia virus reverse transcriptase). The resulting cDNA can be used as a template for a standard PCR.
Nested PCR is a strategy where two pairs of PCR primers (forward and reverse) target a single locus with a B set ‘nested’ within an A set. The initial amplification pair (A set) generates an ~1kb amplicon off (total RNA /RT derivative) cDNA. The second amplification pair (B set; nested pair) target within the first amplicon, and produce a secondary ~500bp amplicon. The probability of reaching specific B set logarithmic amplification is dependent on an initial A set dependent amplification.
A set primer amplicon size ~1000 bp
B set primer amplicon size 250-500 bp
- To identify low levels of DNA contamination, do a PCR of a housekeeping gene and a portion of the RNA preparation as template. If there is contamination, there will be products in all samples.
Primer Tm Values
Tm values for PCR primers range between 55-60 C (19-22 nt, GC% ~55%, no Salt) OR 63-68 C w/salt. The A and B nested primer sets share similar base pair length, GC% and Tm values.
Nested PCR utilizes two pairs of PCR primers for a single locus. The first primer pair A set amplifies within the locus. The second primer pair B set (nested primers) then binds within the ‘A’ amplicon to produce a second nested ‘B’ amplicon.
Reagents
- Reverse Transcriptase
- Deoxynucleotide Mix, dATP, dCPT, dGTP, dTTP, 10 mM each, in sterile double-distilled water, pH 8.5
- Reaction Buffer, 10x conc., 1.05 ml; 100 mM Tris-HCl, 500 mM KCl, pH 8.3 (20°C)
- MgCl2 Stock Solution, 2 x 1.3 ml each 25 mM MgCl2
- Gelatin, 0.05% gelatin (w/v)
- Oligo-p(dT)15 Primer, 0.02 A260 units/µl (0.8 µg/µl)
- RNase Inhibitor, 50 U/µl
- Sterile Water
Oligo (dT)15
Oligo-p(dT)15 Primer is a conventional primer component that binds polyAdenylation tracts on protein coding transcripts (mRNA) as an integral part of a reverse transcription reaction (with reverse transcriptase)
- Oligo-p(dT)15 Primer, 0.02 A260 units/µl (0.8 µg/µl)
- Each vial contains 40 µg in 8 nmol of PBS with < 0.1% sodium azide and 0.1% gelatin.
- Product as 40 ug of lyopholized powder store at -20 C.
- Reconstitute with 50 uL (0.8ug/uL) of double distilled water (also store at -20 C).
- Prepare aliquots of the reconstituted form/ avoid repeated freeze and thaw cycles.
PCR Optimization
- MgCl2 concentrations may vary depending on the template, primer, and dNTP concentrations in the amplification reaction. To optimize conditions, use a MgCl2 titration, generally between 0.5 and 10 mM.
- Primer concentrations may vary; typical final concentrations range from 0.01 to 0.5
- The amount of cDNA utilized in RT-PCR reactions may vary depending on the nature of the RNA template; typically, 5 ul of the cDNA of reverse transcribed total RNA, 20 ul of the cDNA resulting from reverse transcribed poly(A)+ RNA, or 20
- Addition of Gelatin (0.01 mg/ml final) stabilizes Taq DNA polymerase during the PCR reaction, yielding more amplification product.
- RNA quality Assessment
- Proposal: gDNA contamination (relative quantification) is acceptable if gDNA amplifies >5 cycles after the cDNA amplification (>32 fold less template).
DNA Contamination
Non-specific amplicons can arise from primer-dimer formation or unspecific background amplification of genomic (gDNA). Primers toward an existing transcript or RNA entity control for false genomic DNA (gDNA dependent) amplicon detection, when reaction conditions yield true (mRNA dependent) amplicon detection.
- GAPDH
Housekeeping genes for normalization of transcript (gene expression) data can be suitable for gDNA contamination controls in qRT-PCR experiments.
- 18s rRNA
- 18S rRNA is proposed to be predominately absent poly(Adenylated) tail, and therefore should not appear to amplify in an Negative Reverse Transcriptase (-RT) control PCR reaction.
-/+ Reverse Transcriptase
RNA quality (purity and integrity) and quantity are critical to reliable gene expression analysis.
Negative Reverse Transcriptase (-RT) as a control when preparing cDNA from an RNA sample extraction preparation.
Reaction 1 (-RT) = Negative RT Control = RNA (DNase treated) + dNTPS + Random Hexamers/Oligo dT + RT Buffer + Water // NO Reverse Transcriptase//. Reaction 2 (+RT )= WITH Reverse Transcriptase (cDNA from polydt)
Perform PCR with housekeeping primers.
No gDNA contamination / -RT will be negative (no amplicon) / +RT will be positive YES gDNA contamination/ -RT will be positive (yes amplicon); incomplete DNase digestion of the RNA prep.
Performing amplification of a sample prior to reverse transcriptase / minus reverse transcriptase control (no reverse transcriptase) determines to what extent genomic DNA contamination exists from the RNA prep. Determination of DNA contamination present in an RNA preparation controls for false positive qPCR. Amplification of traditional housekeeping genes (ie GAPDH) within a RNA prep (no RT) suggests gDNA contamination.
DNase digestion
DNase digestion, for primers that do not span exon/exon boundaries (ie GAPDH), one proposal is to digest RNA prior to RT (1 µg RNA + 1 unit DNase I, 37°C 45 min).
cDNA Synthesis (Reverse Transcription)
In a standard RT-PCR assay, varying amounts of RNA template 10ug, 1ug, 100ng, 100pg are reversely transcribed with a poly dT primer that attaches to the polyadenylation track on mRNA to yield a cDNA template.
- Prepare a solution containing:
a) 1 ul oligo (dT)12–18 (500 ug/ml)
b) 1 ng-5 ug total RNA
c) 1 ul 10 mM dNTPs
d) and add RNase-free water to a final volume of 12 ul
- If extensive secondary structure is potentially present in the RNA, the RNA sample may be denatured at +70°C for 5 min before adding it to the reaction minimize RNA secondary structure, and placed on ice for 5 min before adding it to the reaction.
a) 4 ul 5x reverse transcriptase buffer
b) 2 ul 0.1 M DTT
c) 1 u RNase inhibitor
Reverse Transcritpion Reaction
- Incubate at +30°C for 10 min minutes to anneal primer and template.
- Add 1 ul reverse transcriptase (200 units) and incubate at 42° C for 60 minutes to extend the primer and then terminate the reaction by incubating at 70° C for 15 minutes. The RNA is subsequently reverse transcribed, resulting in cDNA synthesis.
NOTE: (As an optional step add 1 ul RNase H (2 unit/ul) and incubate at 37° C for 20 minutes)
First PCR reaction
- Prepare a solution containing:
a) 5 ul 10x PCR buffer (with or without MgCl2)
b) 5 ul 25 mM MgCl2 (It may be necessary to vary the MgCl2 concentration, 2.5 mM final concentration recommended.)
c) 1 ul 10 mM dNTP
d) 1 ul primer pair A
e) 1 ul Taq DNA polymerase
f) 2 ul cDNA and add water to 50 ul
First PCR amplification parameters
Perform 15–35 cycles of PCR. Annealing and extension conditions are primer and template dependent and must be determined empirically for each template-primer pair. Tm values for PCR primers generally range between 55-60 C
- Denaturation 96°C, 0.5-1.5 minute
- Annealing 57°C, 0.5-1 minute
- Polymerization (35 cycles) 72°C, 0.5-2 minutes
- Link; Extension (1 cycle) 70°C, 5 minutes
- Link to a 4°C Soak file.
NOTE: Addition of Gelatin (0.01 mg/ml final) stabilizes Taq DNA polymerase, yielding more amplification product.
Second (nested) PCR reaction
- Prepare a solution containing:
a) 5 ul 10x PCR buffer (with or without MgCl2)
b) 5 ul 25 mM MgCl2 (It may be necessary to vary the MgCl2 concentration, 2.5 mM final concentration recommended)
c) 1 ul 10 mM dNTP
d) 1 ul primer pair B
e) 1 ul Taq DNA polymerase
f) 1–5 ul first PCR product and add water to 50 ul
Second PCR amplification parameters
Perform 15-25 cycles of PCR. Annealing and extension conditions are primer and template dependent and must be determined empirically for each template-primer pair. Tm values for PCR primers generally range between 55-60 C
- Denaturation 96°C, 30 sec
- Annealing 57°C, 30 sec
- Polymerization (35 cycles) 72°C, 30 sec
- Link; Extension (1 cycle) 70°C, 5 minutes
- Link to a 4°C Soak file.
NOTE: Addition of Gelatin (0.01 mg/ml final) stabilizes Taq DNA polymerase, yielding more amplification product.
- PCR products are visualized by agarose gel electrophoresis stained with an appropriate dye.
Agarose Gel Electrophoresis
Agarose gel electrophoresis can resolve DNA or RNA by size. DNA/RNA is visible in the gel when ethidium bromide is added during the gel casting process. EtBr intercalates between dsDNA or dsRNA and absorbs invisible UV light and emits visible orange light. SYBR Green absorbs blue light (λmax = 488 nm) and emits green light (λmax = 522 nm) may be used in place of EtBr, although more commonly this dye is used for qPCR.
- 0.7% gel will show good separation (resolution) of large DNA fragments (5–10kb)
- 2% gel will show good resolution for small fragments (0.2–1kb)
- 3% gel or vertical polyacrylamide gel for separating smaller fragments (the higher the % gel, the more brittle the gel)
Gel Casting
The volume of agarose required for a minigel is around 30–50mL.
- Weigh out 0.5g of agarose into a 250mL conical flask. Add 50mL of 0.5xTBE, swirl to mix.
- Microwave for about 1 minute to dissolve the agarose. Use gloves to handle and do not let it overboil; molten agarose is very hot.
- Let cool for 5 minutes or warm to touch with bare hands.
- Add 1µL of ethidium bromide (10mg/mL) and swirl to mix.
- Pour the gel into the resevoir. Push any bubbles away to the side using a disposable tip. Insert the comb. Wait at least 30-60 min.
- Add 0.5x TBE buffer into the gel tank to submerge the gel to 2–5mm depth. This is the running buffer.
- Load samples and run the gel no greater than 5 Volt/cm.
2L of 10xTBE
- 218g Tris base
- 110g Boric acid
- 9.3g EDTA
Dissolve the ingredients in 1.9L of distilled water. pH to about 8.3 using NaOH and make up to 2L.
Loading buffer
- 25mg bromophenol blue or xylene cyanol (Bromophenol blue migrates @ ~200–400bp. Xylene cyanol migrates ~4kb)
- 4g sucrose
- H2O to 10mL
Store at 4°C or -20
RNA extraction-PDF

Comparison of purification approaches
Guanidine isothiocyanate extraction coupled with lithium chloride precipitation
- Lyses cells, extracts cellular RNA and denatures proteins all at once in many cases.
- Inactivates RNases faster than acid phenol extraction.
- Does not physically separate RNA from proteins and DNA in one step. Protein contamination must be removed by chloroform treatment and need to differentiate between RNA and DNA using some other technique (like lithium chloride precipitation).
- Can also use cesium chloride ultra-centrifugation instead of lithium chloride precipitation but this requires access to an ultra-centrifuge.
Acid phenol extraction and alcohol precipitation
- Very cheap
- Long procedure
- Prone to DNA contamination
- Can leave residual phenol in the sample inhibiting downstream reactions and introducing error into RNA quantitation.
- Doesn’t inactivate RNases immediately.
TRIzol or tri followed by chloroform and precipitation
- trizol or tri (name depends on manufacturer) combines phenol and guanidine isothiocyanate and thereby some of the advantages of the above two
- removes protein and DNA but depends on pipetting skills (disturbing the phases leads to contamination)
- RNA is protected by the reagent during the extraction procedure
- phenol and chloroform are potentially harmful reagents (handle under the hood)
For detailed protocols see:
- RNA extraction using trizol/tri
- Trizol extraction for RNA and DNA
Anion-exchange matrices
- Allows purification of genomic DNA and RNA in parallel.
Silica matrices
- Very small species (< 200nt) do not bind to silica matrices (except for Norgen columns which get miRNAs).
Commercially available kits
- Qiagen RNeasy kits (silica column tubes) are the most widely recommended. It is more expensive than other kits but is good for transcriptomics.
- The column binding system of RNAeasy is faster and without harmful chemical but it has a lower maximum yield (due to the binding capacity of the columns) and will remove RNA smaller than ~200 nt.
- All the kits are suitable for applications like RT-PCR as long as you check for and remove contaminants from RNA preparation.
Harvesting cells
- Centrifugation of cells can induce a stress response in cells altering transcript levels (potentially unequally across different transcripts).
- “RNAprotect” is a reagent used to stabilize RNA content in bacterial cells grown in liquid culture.
- Works best if cells are grown in minimal media. Works worst in LB liquid culture due to complex components.
- Prepare all reagents and organize all equipment ahead of time to minimize the time between end of cell growth and cell lysis (especially if stabilization reagents are not used).
- Once cells are lysed, keep all samples on ice and use ice cold reagents to reduce RNase activity. However, note that RNases are still active at 0°C. (Do not put cells on ice prior to cell lysis otherwise you risk inducing a cold-shock response.)
Lysing cells
- Cell lysis is the point at which things are most likely to go wrong.
- Mechanical disruption and homogenization can help with cell lysis issues. Using a bead grinding machine may be helpful.
Bacteria
- The best way to lyse bacterial cells is guanidine isothiocyanate which isolates cellular RNA at the same time.
- Pre-digestion of the cell wall may improve lysis efficiency. This is essential for Gram-positive bacteria. Use 0.4mg/mL lysozyme for Gram-negative bacteria and 3mg/mL lysozyme for Gram-positive bacteria in chosen resuspension buffer.
- Use 2mL of lysozyme containing buffer per 10mL of E. coli bacterial culture having an OD600nm of 0.5 (assumes OD600nm of 1 equals 109 cells per mL). Scale the amount of resuspension buffer linearly based on the number of cells to be lysed.
- To lyse cells, add resuspension buffer and pipette up and down rapidly using a 250 μL capacity pipette tip until no cell clumps are visible. Incubate on the bench for 5 mins (Gram-negative bacteria) or 15 mins (Gram-positive bacteria). (This lysis procedure assumes that RNA stabilization reagents have been used or that resuspension buffer contains RNase inhibitors.)
- Homogenization can improve efficiency of RNA isolation from bacterial cells. If cell lysate is viscous, pour lysate into a syringe and pass the solution back and forth through a 20 gauge needle 5-10 times.
RNA separation from other cellular molecules
Several different possibilities exist to purify RNA from complete cell lysate (see the comparison above). For mammalian cell culture and tissues trizol/tri and silica columns (e.g. RNAeasy) are probably the most common.
- Main article: RNA extraction using trizol/tri
Solubilization of RNA
- To dissolve RNA pellets, use RNase-free water (which is least likely to interfere with downstream applications).
- If required heat the RNA After adding water to promote solubilization (70°C for 5 mins, mix by pipetting every minute).
- RNA at a concentration of 1 μg/μL is too dilute to re-precipitate upon cooling.
Quantification
RNA is typically quantified spectrometrically using absorption at 260nm (A260). An A260 reading of 1 is equivalent to ~40 µg/ml of single-stranded RNA [1]. Machines that require very little sample volume for testing like NanoDrop are popular.
- Absorption measurement depends on the buffer (use the same buffer as blank)
| diluent | A260/A280 |
|---|---|
| DEPC water (pH 5-6) | 1.60 |
| Nuclease-free water (pH 6-7) | 1.85 |
| TE (pH 8.0) | 2.14 |
- Depending on machine the A260 reading of the RNA sample should be between 0.1-0.5. Dilute the sample in H2O for this measurement.
- NanoDrop’s upper limit for RNA concentration is 3000 µg/µl; dilute if necessary
- absorption is highly pH dependent (see also table above and PMID 9067025)
Storage
- Store RNA at less than -70°C. (why?)
- Only freeze-thaw RNA samples once. (freeze thaw as little as possible)
- Re-determine RNA concentration following storage and defrosting.
Sources of contamination
DNA
- To identify low levels of DNA contamination, do a PCR of a housekeeping gene and a portion of the RNA preparation as template. If there is contamination, there will be products in all samples.
- Use lithium chloride re-precipitation to remove DNA for best results. However, this is slow.
- Use DNaseI if you are in a hurry.
Protein
- Measure th A260/A280 ratio. It should be 2.0 for very pure RNA samples. When measuring this ratio, dilute the RNA sample in TE buffer (not H2O) because pH can affect the A280 reading.
- If there is protein contamination, you may need to do a chloroform cleaning and reprecipitation.
Salt
- Measure the A260/A240 ratio. It should be 1.4 for very pure RNA samples.
- Isopropanol precipitation coupled with a 70% v/v ethanol wash of the pellet can remove salts.
Supplemented M9 broth (M9sup)-PDF
Materials
| 50 mL | 500 mL | 1000 mL | |
|---|---|---|---|
| dH2O | 43 ml | 430 ml | 860 ml |
| 10 X M9 salts + glucose | 5 ml | 50 ml | 100 ml |
| 10% Casamino acids | 1 ml | 10 ml | 20 ml |
| 1% Thiamine | 50 µl | 0.5 ml | 1 ml |
| 1 M MgSO4 | 100 µl | 1 ml | 2 ml |
| 1 M CaCl2 | 5 µl | 50 µl | 0.1 ml |
Procedure
Pre-sterilize all component solutions and aseptically add to sterile water. Alternatively, mix all components and then filter-sterilize through a 0.22 µm filter. Store at 4 ºC.
Notes
Please feel free to post comments, questions, or improvements to this protocol. Happy to have your input!
Making frozen permanents-PDF
Materials
- Cryo vials
- Overnight bacterial culture
- LB (50% glycerol)
Procedure
Pipette 400 μL LB 50% glycerol and 600 μL overnight culture into a cryo vial. Vortex, put in a freezer box and store in the -80 degree freezer.
Notes
- When making LB in 50% glycerol, measure out the glycerol in a graduated cylinder before adding water. To get the last of the glycerol out of the cylinder, add the required amount of water, cover the top with parafilm, and shake before pouring out.
Digestion-PDF
Overview
Standard DNA single or double digest of variable volume V using NEB restriction enzymes.
Materials
- X μL DNA template
- 10X NEB Digestion Buffer Y (Y = 1, 2, 3, or 4)
- 10mg/ml BSA
Procedure
- Determine compatibility and reaction conditions using Double Digest Finder (double digest only).
- Determine reaction volume V by computing V = [(X + 1)/0.8] μL (single digest) or V = [(X + 2)/0.8] μL (double digest).
- To DNA sample, add
- 0.1V 10X NEB Buffer Y (Y = 1, 2, 3, or 4)
- 0.1V 10mg/ml BSA
- 1 μL restriction enzyme 1
- 1 μL restriction enzyme 2 (double digest only)
- Incubate 3 hr to overnight at reaction temperature given by NEB (standard = 37 °C).
- Optional: Heat inactivate restriction enzyme(s) by incubation 80 °C 20 min. Not necessary when digestion product is going to be purified or gel extracted, as these procedures remove the enzymes.
- Lab standard double digestion uses X = 22 μL plasmid DNA, so V = (22 + 2)/0.8 μL = 30 μL.
- 22 μL plasmid DNA
- 3 μL 10X NEB Buffer Y
- 3 μL 10mg/ml BSA
- 1 μL restriction enzyme 1
- 1 μL restriction enzyme 2
Electrocompetent cells-PDF

Materials
- GYT (glycerol, yeast extract, tryptone)
- • 10%(v/v) glycerol
- • 0.125% (w/v) yeast extract
- • 0.25% (w/v) tryptone
- DI water
- 10% Glycerol
Special Equipment
- Centrifuge
- Ice water bath
- Liquid nitrogen
Method
Important: All steps in this protocol should be carried out aseptically
- Inoculate: Prepare flask containing 50 ml of LB medium. Pick up a single colony of cells from plate (using a sterile toothpick) and swirl around inside flask. Incubate the culture overnight at 37°C with vigorous aeration (250 pm in a rotary shaker).
- Dilute and incubate: Inoculate two aliquots of 475 ml of prewarmed LB medium in separate 2-liter flasks with 25 ml of the overnight bacterial culture. Incubate the flasks at 37°C with agitation (300 cycles/min in a rotary shaker). Measure the OD-600 every twenty minutes (this step will take around 1.5-2 hrs).
- Rapidly cool culture: Once the OD-600 of the culture reaches 0.6-1.0 (recommends 0.4), rapidly transfer the flasks to the pre-made ice-water bath for 15-30 minutes. Swirl the culture occasionally to ensure that cooling occurs evenly. In preparation for the next step, place the centrifuge bottles in the ice-water bath as well.
Note: After this point, do not let your cells warm up past 4°C
Note: When harvesting cells by decanting, be very careful to not disturb the pellet– this could result in a much lower yield. If necessary, aspirate instead of decant the supernatant. Get someone to show you how to aspirate. Also, if the pellet seems loose, sometimes it is helpful to re-spin the cells down.
- Centrifuge 1: Transfer the cultures to ice-cold centrifuge bottles. Harvest the cells by centrifugation at 1000g (2500 rpm) for 15 minutes at 4°C. Decant the supernantant and resuspend the cell pellet in 500 ml of ice-cold DI water. Note: I think this should be done for each of the two 500ml cultures, i.e this is a 1:1 resuspension rather than a concentration by a factor of 2 .
- Centrifuge 2 (water): Harvest the cells by centrifugation at 1000g for 20 minutes at 4°C. Decant the supernatant and resuspend the cell pellet in 250 ml ice-cold DI water.
- Centrifuge 3 (water): Harvest the cells by centrifugation at 1000g for 20 minutes at 4°C. Decant the supernatant and resuspend the cell pellet in 10 ml ice-cold 10% glycerol.
- To spin down your pellet in 10 ml, it might be helpful to use 15-ml Falcon tubes instead of the round-bottom tubes.
- Centrifuge 4 (glycerol): Harvest the cells by centrifugation at 1000g for 20 minutes at 4°C. Carefully decant the supernatant and use a Pastteur pipette attached to a vacuum line to remove any remaining drops of buffer.
- Resuspend in GYT: Resuspend in 1 ml ice cold GYT. This is best done by gently swirling rather pipetting or vortexing.
- Measure OD: Measure the OD-600 of a 1:100 dilution of the cell suspension. (In the cuvette, mix 0.99 mL water and 0.01 mL cell suspension).
Note: The desired concentration is [math]\displaystyle{ 2.5 \times 10^{11} }[/math] cells per mL, giving [math]\displaystyle{ 1\times 10^{10} }[/math] cells per 40 [math]\displaystyle{ \mu }[/math]L. This corresponds to an OD-600 (after 100x dilution) of roughly 3.75. It is difficult to reach this value, but it is still important to know the concentration of cells to calculate efficiencies.
- Dilute the cell suspension to a concentration of 2 x 10^10 to 3 x 10^10 cells/ml (1.0 OD600 = approx. 2.5 x 10^8 cells/ml) with ice-cold GYT medium.
- Test for arcing: Transfer 40 ul of the suspension to an ice-cold electroporation cuvette (0.1-0.2 cm gap, on middle shelf next to electroporator) and test whether arcing occurs when an electrical discharge is applied. Place the cuvette in the green holder attached to the machine. Go to option 4, Pre-set protocols; choose bacterial; choose the correct choice for your size cuvette, probably the first option for a .1 cm cuvette. If arcing occurs, wash the remainder of the cell suspension once more with ice-cold GYT medium to ensure that the conductivity of the bacterial suspension is sufficiently low (<5 mEq).
- Storage: Store cells at -80°C until they are required for use. For storage, dispense 40 ul aliquots of the cell suspension into sterile, ice-cold .5 ml microcentrifuge tubes, drop into a bath of liquid nitrogen and transfer to a -80°C freezer. To remove the tubes from the liquid nitrogen bath, bring out into the hall along with a storage box, and pour the tubes and liquid nitrogen into the box. Once all the tubes are out, close the box most of theh way and let the liquid run out into the hallway. Try not to do this in the very center of the walkway!
To use frozen cells: Remove an appropriate number of aliquots of cells from the -80°C freezer. Thaw the tubes on ice.
Chung cells-PDF
Materials
- Overnight E. coli culture
- 50 mL Falcon tubes
- LB
- TSS
- 1.5 mL centrifuge tubes
Storage:
- Freezer box
Transformation:
- LB or TSS with 20 mM glucose
- Plasmid DNA
Procedure
- Dilute an overnight culture of E. coli 1:100 in LB media. Grow to an OD of 0.3 and put the culture on ice.
- Transfer the culture to chilled 50 mL Falcon tubes and centrifuge at 5,000 g for 10 minutes at 5°C.
- Discard the supernatant and resuspend at 1/10th volume with cold TSS by gently pipetting up and down.
- Transfer 100 μL aliquots to chilled 1.5 mL centrifuge tubes, and either use immediately or freeze in liquid nitrogen and store in the -80 freezer.
- To tranform the cells, add 1-3 μL prepped plasmid DNA to a 100 μL aliquot of Chung cells (first thaw on ice if frozen), mix gently, and store at 5°C for 5-60 minutes. Don’t forget to set up a control using sterile water instead of plasmid DNA.
- Add 900 μL of either SOC or LB with 20 mM glucose, and incubate the culture with shaking for one hour at 37°C (larger vectors may require a longer recovery period).
- Plate 10 μL, 100 μL, and the rest of the transformation on selective media.
Notes
Please feel free to post comments, questions, or improvements to this protocol. Happy to have your input!
- This protocol is adapted from Chung and Miller (1989). One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci., 86(April), 2172–2175.
- When making TSS, first add PEG to LB, heat in a 50°C water bath, and then swirl to mix. Add Mg2+, wait until the solution gets to room temperature before adding DMSO, and filter sterilize. Store in the 5 degree fridge.
Antigen retrieval-PDF
Detection techniques using antibodies often fail to work on PFA- or formalin-fixed, paraffin-embedded sections. Antigen retrieval methods can then, in some cases, enable specific antibody detection. They work by reversing some of the chemical modification of epitopes during fixation. These procedures will not help much with epitope loss due to denaturation during sample treatment, like hot paraffin embedding.
Types of retrieval methods
Heat treatment
- Mechanism: heat cleaves cross-links, exposes buried epitopes of proteins, protein unfolding/refolding
- Specific protocol 1: citrate buffer
- Specific protocol 2: Tris-EDTA buffer
Protease treatment
- Specific protocol 1: proteinase K
- Specific protocol 2: trypsin
Detergent treatment
- Specific protocol: SDS