Affymetrix Eukaryotic Gene Expression Sample Processing
This protocol will allow you to perform the hybridization, washing, staining, and scanning of Affymetrix GeneChip microarrays. This protocol is a supplement to instructions provided in the Affymetrix Expression Manual.
Workflow
Materials
List reagents, supplies, and equipment necessary to perform the protocol here. For those materials which have their own OWW pages, link to that page. Alternatively, links to the suppliers’ page on that material are also appropriate.
- Hybridisation controls: These are part of the complete One-Cycle Target Labeling and Control Reagents, Affymetrix, P/N 900493; and may be ordered separately if necessary Usually not!)
- GeneChip Eukaryotic Hybridization Control Kit, Affymetrix, P/N 900454 (30 reactions)
- GeneChip Eukaryotic Hybridization Control Kit, Affymetrix, P/N 900457 (150 reactions)
- Control Oligo B2, 3 nM, Affymetrix, P/N 900301
Other reagents:
- Water, Molecular Biology Grade, BioWhittaker Molecular Applications / Cambrex, P/N 51200
- Bovine Serum Albumin (BSA) solution (50 mg/mL), Invitrogen Life Technologies, P/N 15561-020
- Herring Sperm DNA, Promega Corporation, P/N D1811
- 5M NaCl, RNase-free, DNase-free, Ambion, P/N 9760G
- MES hydrate SigmaUltra, Sigma-Aldrich, P/N M5287
- MES Sodium Salt, Sigma-Aldrich, P/N M5057
- EDTA Disodium Salt, 0.5M solution (100 mL), Sigma-Aldrich, P/N E7889
- DMSO, Sigma-Aldrich, P/N D5879
- Surfact-Amps 20 (Tween-20), 10%, Pierce Chemical, P/N 28320
- R-Phycoerythrin Streptavidin, Molecular Probes, P/N S-866
- 5M NaCl, RNase-free, DNase-free, Ambion, P/N 9760G
- PBS, pH 7.2, Invitrogen Life Technologies, P/N 20012-027
- 20X SSPE (3M NaCl, 0.2M NaH2PO4, 0.02M EDTA), BioWhittaker Molecular Applications / Cambrex, P/N 51214
- Goat IgG, Reagent Grade, Sigma-Aldrich, P/N I 5256
- Anti-streptavidin antibody (goat), biotinylated, Vector Laboratories, P/N BA-0500
- Tygon Tubing, 0.04″ inner diameter, Cole-Parmer, P/N H-06418-04
- Tough-Spots, Label Dots, USA Scientific, P/N 9185-0000
Reagent Preparation
- 12X MES Stock Buffer (1.22M MES, 0.89M [Na+]) 250mL
- 16.15g of MES hydrate
- 48.325g of MES Sodium Salt
- 200 mL of Molecular Biology Grade water
- Mix and when fully dissolved adjust volume to 250 mL. The pH should be between 6.5 and 6.7. Filter through a 0.2 µm filter. Do not autoclave. Store at 2°C to 8°C and shield from light. Discard solution if yellow.
- 2X Hybridization Buffer (Final 1X concentration is 100 mM MES, 1M [Na+], 20 mM EDTA, 0.01% Tween-20) 50 mL
- 8.3 mL of 12X MES Stock Buffer
- 17.7 mL of 5M NaCl
- 4.0 mL of 0.5M EDTA
- 0.1 mL of 10% Tween-20
- 19.9 mL of water
- Do not autoclave. Store at 2°C to 8°C, and shield from light. Discard solution if yellow.
- 1X Hybridization Buffer (100 mM MES, 1M [Na+], 20 mM EDTA, 0.01% Tween-20) 50 mL:
- 25 mL of 2X Hybridization Buffer
- 25 mL of water
- Wash Buffer A: Non-Stringent Wash Buffer (6X SSPE, 0.01% Tween-20) 1,000 mL:
- 300 mL of 20X SSPE
- 1.0 mL of 10% Tween-20
- 699 mL of water
- Filter through a 0.2 µm filter
- Wash Buffer B: Stringent Wash Buffer (100 mM MES, 0.1M [Na+], 0.01% Tween-20) 250 mL:
- 20.825 mL of 12X MES Stock Buffer
- 1.3 mL of 5M NaCl
- 0.25 mL of 10% Tween-20
- 227.625 mL of water
- Filter through a 0.2 µm filter
- Store at 2°C to 8°C and shield from light. Keep this in the bottle shielded with foil during use.
- 2X Stain Buffer (Final 1X concentration: 100 mM MES, 1M [Na+], 0.05% Tween-20) 250 mL:
- 41.7 mL of 12X MES Stock Buffer
- 92.5 mL of 5M NaCl
- 2.5 mL of 10% Tween-20
- 113.3 mL of water
- Filter through a 0.2 µm filter
- Store at 2°C to 8°C and shield from light
- 10 mg/mL Goat IgG Stock
- Resuspend 50 mg in 5 mL of 150 mM NaCl
- Store at 4°C
- If a larger volume of the 10 mg/mL IgG stock is prepared, aliquot and store at -20°C until use. After the solution has been thawed it should be stored at 4°C. Avoid additional freezing and thawing.
Protocol
These protocols provide detailed steps for preparing the eukaryotic hybridization mix containing labeled target and control cRNA. After completing the procedures described in this chapter, the hybridized probe array is ready for washing, staining, and scanning, as detailed in Washing, Staining and Scanning GeneChip Arrays.
Notes before starting
- It is imperative that frozen stocks of 20X GeneChip Eukaryotic Hybridization Controls are heated to 65°C for 5 minutes to completely resuspend the cRNA before aliquotting.
- It is important to allow the arrays to equilibrate to room temperature completely. Specifically, if the rubber septa are not equilibrated to room temperature, they may be prone to cracking, which can lead to leaks.
- Eukaryotic hybridisations are generally done at 45°C.
- Streptomyces hybridisations are done at 50°C. CHECK PACKAGE INSERTS!
Setting up hyb cocktail
Please refer to the table below for the necessary amount of cRNA required for appropriate probe array format. These recipes take into account that it is necessary to make extra hybridization cocktail due to a small loss of volume (10-20 µL) during hybridization.
- Make a master mix of hybridisation cocktail reagents and add to individual fragmented cRNAs in 1.5ml tubes.
- If using the GeneChip IVT Labeling Kit to prepare the target, a final concentration of 10% DMSO needs to be added in the hybridization cocktail for optimal results.
Physical preparation of the array hybridisation
- Equilibrate probe array to room temperature immediately before use.
- Heat the hybridization cocktail to 99°C for 5 minutes in a thermomixer.
- Wet the array by filling it through one of the septa (see below) with 1X Hybridization Buffer using a pipette and appropriate tips.
It is necessary to use two pipette tips when filling the probe array cartridge: one for filling and the second to allow venting of air from the hybridization chamber.
- Incubate the probe array filled with 1X Hybridization Buffer at 45°C (50°C for Streptomyces) for 10 minutes with rotation at 60rpm.
- Transfer the hybridization cocktail that has been heated at 99°C, to a 45°C (50°C for Streptomyces) thermomixer for 5 minutes.
- Spin hybridization cocktail(s) at maximum speed in a microcentrifuge for 5 minutes to remove any insoluble material from the hybridization mixture.
- Remove the buffer solution from the probe array cartridge.
- Fill with appropriate volume of the clarified hybridization cocktail, avoiding any insoluble matter at the bottom of the tube. Label the array with the JobID_SampleID!
- Place probe array into the Hybridization Oven, set to 45°C (50°C for Streptomyces). Avoid stress to the motor; load probe arrays in a balanced configuration around the axis. Rotate at 60rpm.
- Hybridize for 16 hours. During the latter part of the 16-hour hybridization, proceed to Washing, Staining, and Scanning and prepare reagents required immediately after completion of hybridization.
Washing, Staining, and Scanning
These protocols provide detailed instructions for using the Fluidics Station 450 and GeneChip Scanner 3000 to automate the washing, staining and scanning of GeneChip expression probe arrays. After completing the procedures described in this chapter, the scanned probe array image (.dat file) is ready for analysis. After 16 hours of hybridization, remove, and keep, the hybridization cocktail from the four arrays you are washing and fill completely with Non-Stringent Wash Buffer (Wash Buffer A), as given the table at step 1 of Eukaryotic Target Hybridization. This procedure takes approximately 90 minutes to complete.
Step 1: Starting up workstation, fluidics and scanner. Turn on PC and after you have logged in start GCOS software. Turn on the fluidics station using the toggle switch on the lower left side of the machine.. Turn on the scanner (it is recommended to wait until nearly all your arrays are ready for scanning. The autoloader should allow you to walk away and the scanner does make quite a lot of noise).
Step 2: Entering Experiment Information To wash, stain, or scan a probe array, an experiment must first be registered in GCOS. The fields of information required for registering experiments in GCOS are: Sample Name: user-supplied-sample-name_JobID_SampleID_s e.g. Col1_39_7_s Sample Type Project Experiment Name: same as above without _s e.g. Col1_39_7 Probe Array Type
You should also enter the following fields if possible: Barcode – Scan in the barcodes when possible as this allows GCOS to attach a .dat file to the experiment.
The Project, Sample Name, and Experiment Name fields establish a sample hierarchy that organizes GeneChip gene expression data in GCOS. In terms of the organizational structure, the Project is at the top of the hierarchy, followed by Sample Name, and then Experiment Name.
Step 3: Preparing the Fluidics Station The Fluidics Station 450 is used to wash and stain the probe arrays. It is operated using GCOS. Setting Up the Fluidics Station Turn on the Fluidics Station if you have not already done so. Place Wash A and Wash B buffers on the fluidics station and make sure tubing is all the way inside the bottle so no air will be sucked up. Wash B is stored in the fridge and should be kept as dark as possible during use. Make sure there is enough buffer in each bottle for the number of chips you are going to run. Select Run → Fluidics from the menu bar. Or click the fluidics button on the toolbar; you will be presented with the fluidics station window (see below).
Priming the Fluidics Station Priming ensures that the lines of the fluidics station are filled with the appropriate buffers and the fluidics station is ready for running fluidics station protocols. Priming should be done: when the fluidics station is first started. when wash solutions are changed. before washing, if a shutdown has been performed. if the LCD window instructs the user to prime.
Select the Prime_450 protocol and choose All Modules, then click the Run button (this can be run on individual modules if you are only running one or two arrays.)
Step 4: Preparing the Staining Reagents Prepare the following reagents. Volumes given are sufficient for one probe array. Always prepare the stain solutions fresh, on the day of use.
Streptavidin Phycoerythrin (SAPE) should be stored in the dark at 4°C, foil-wrapped. Mix SAPE well before preparing stain solution. Do not freeze SAPE.
SAPE Solution Mix
Master Mix 2X Stain Buffer 600.0 µL 50 mg/mL BSA 48.0 µL 1 mg/mL Streptavidin Phycoerythrin (SAPE) 12.0 µL DI H20 540.0 µL
Antibody Solution Mix
Master Mix 2X Stain Buffer 300.0 µL 50 mg/mL BSA 24.0 µL 10 mg/mL Goat IgG Stock 6.0 µL 0.5 mg/mL biotinylated antibody 3.6 µL DI H20 266.4 µL
Aliquot stain solutions into tubes marked 1 & 3 (SAPE Stain) and 2 (Antibody) with 600 µL in each tube for use on the fluidics station.
Step 5: Washing and Staining the Probe Array using the Fluidics450 1. In the Fluidics Station dialog box in GCOS, select the correct experiment name from the drop-down Experiment list. (The Probe Array Type appears automatically, check that it corresponds to the array type you are using.) 2. Select the appropriate protocol from the drop-down Protocol list, to control the washing and staining of the probe array format being used: Check array package insert for correct fluidics protocol to use. 3. Choose Run in the Fluidics Station dialog box to begin the washing and staining. Follow the instructions in the LCD windows on the fluidics station modules. 4. Insert the appropriate probe array into the designated module of the fluidics station while the cartridge lever is in the down, or eject position. When finished, verify that the cartridge lever is returned to the up, or engaged, position. 5. Remove any microcentrifuge vial remaining in the sample holder of the fluidics station module(s) being used. 6. When prompted “Load Vials 1-2-3,” place the three experiment sample vials (the microcentrifuge vials) into the sample holders 1, 2, and 3 on the fluidics station. Place one vial containing 600 µL of SAPE solution in sample holder 1. Place one vial containing 600 µL of antibody solution in sample holder 2. Place one vial containing 600 µL of SAPE solution in sample holder 3. Press down on the needle lever to snap needles into position and to start the run. The Fluidics Station dialog box at the workstation terminal and the LCD window display the status of the washing and staining progress. Each run takes about 90 minutes. 7. When prompted “eject cartridge”, remove the array and check for air bubbles. If bubbles are present they need to be removed prior to scanning. Follow instructions on Removing Air Bubbles Before Scanning. 8. When prompted “remove vials”, replace the microcentrifuge vials containing stain solutions with three clean empty microcentrifuge vials. The fluidics station will now take about 20 minutes to complete the protocol and then you are ready to start the next four chips.
Remove and bubbles from the arrays. (See optional step next.)
If you do not scan the arrays immediately, store them at 4°C and in the dark until ready for scanning. Arrays can be stored for 24hours after washing and staining is complete.
When you have finished all array processing you must run the shutdown protocol with all three reagent lines in water.
Once a month you should run the Bleach cleanup protocol, detailed in the Affymetrix manual.
Step optional: Removing Air Bubbles Before Scanning. You must remove any air bubbles before scanning. The easiest way is to insert a 200 µLtip into the top septa and use another 200 µL tip to remove about half of the wash A inside the array. Still holding the pipette in the same position, remove the tip and fill with new wash A, insert it back into the lower septa and fill the array making sure you do not introduce a new bubble.
Probe Array Scan
The scanner is controlled by GCOS. The array is scanned after the wash protocols are complete. Make sure the laser is warmed up prior to scanning by turning it on at least 10 minutes before use. If probe array was stored at 4°C, it will need to warm to room temperature before scanning; a dialogue box will prompt for this when you start any scan.
Handling the GeneChip Probe Array before a Scan If necessary, before you scan the probe array, clean the glass surface with a non-abrasive towel or tissue. Do not use alcohol to clean glass. Before scanning the probe array cartridge, apply Tough-Spots™ to each of the two septa on the probe array cartridge to prevent the leaking of fluids from the cartridge during scanning.
1. On the back of the probe array cartridge, clean excess fluid from around septa. 2. Carefully apply one Tough-Spot to each of the two septa. Press to ensure that the spots remain flat. If the Tough-Spots do not apply smoothly, that is, if you observe bumps, bubbles, tears, or curled edges, do not attempt to smooth out the spot. Remove the spot and apply a new spot. The scanner uses a laser and is equipped with a safety interlock system. Defeating the interlock system may result in exposure to hazardous laser light. You must have read, and be familiar with, the operation of the scanner before attempting to scan a probe array.
Scanning the Probe Array The Scanner 3000 is fitted with an autoloader. This is cooled and must be loaded from position 1 which is outlined in red. Chips can only be loaded in the correct orientation. 1. Select Run → Scanner from the menu bar. Alternatively, click the Start Scan icon in the tool bar. The Scanner dialog box appears with a drop-down list of experiments that have not been run. 2. Verify whether the arrays are at room temperature or not. 3. Click the Start button. 4. Each array will take about 5 minutes to scan. The autoloader and GCOS should allow you to walk away as the scan is organised by the barcode on the arrays.
If you need to the scanner can be operated in manual mode with or without the autoloader. Refer to the scanner Quick Reference Card.
Affymetrix data analysis and formatting.
GCOS analysis
Data needs to be analysed in GCOS and the formatted for QC analysis and customer use.
- Check analysis settings for the array type you are analyzing.
Scaling should be ‘All Probe Sets’ and ‘Target Signal’ should be 100.
- Select the .cel files you want to analyse, right click and select ‘Analyze’.
- Save the pivot data table as a tab delimited txt file with the customer name_JobID.
- Select the .chp files you need QC reports for, right click and select ‘Report’.
- Copy the .txt file and .rpt files for the job from D:\Program Files\Affymetrix\GeneChip\Affy_Data\Data to \\Jigenomelab\affymetrix data\GCOSExport.
- The LIMS should now present rpt files for tech QC analysis.
- Check that all samples are broadly similar to each other, and usually to the chip average.
- Check SF (Scaling Factors) are below 3.
- Check %P (Percent Present) is around 50-65%
- Check Housekeeping controls and Spike in controls 3’:5’ ratios.
- The LIMs should allow customers to download their pivot data txt file.
Affymetrix data backup and formating.
Data needs to be backed up using Affymetrix Data Transfer Tool and formatted for customer use with other analysis packages, i.e. cel file analysis.
DTT Backup file creation
- Open Data Transfer Tool
- Choose the transfer settings: DTT Archive File (No Span), click OK. Click next.
- Use the project filter to find the project you are working on. Click next.
- Highlight the project. Check that the ‘Step2’ checkbox for DAT files is selected. Set ‘Save Location’ to F:\GenomeLab on Tera\Microarray\Affymetrix DTT Backups. Set ‘File Name’ to CustomerName_JobID_ADTT. Click ‘Review’.
- Click start.
- Transfer the ADTT files to DVD at regular intervals for secure backup.
CEL Flat file export
- Open Data Transfer Tool
- Choose the transfer settings: Flat Files (XML-DAT-CEL-CHP), click OK. Click next.
- Use the project filter to find the project you are working on. Click next.
- Highlight the project. Set ‘Save Location’ to C:\AffyBackupData\FlatFiles. Click ‘Review’.
- Click start.
- Move the .CEL and .XML files to a folder with the CustomerName_JobID as the folder name. Delete all remaining Flat Files.
- These files are ready for delivery or collection. You cannot email these files as they are too large.
Notes
- List troubleshooting tips here.
- You can also link to FAQs/tips provided by other sources such as the manufacturer or other websites.
- Anecdotal observations that might be of use to others can also be posted here.
Please sign your name to your note by adding (”’~~~~”’) to the end of your tip.

 Cell lysis only takes a few minutes per well, but tissue homogenisation can take 10-20 minutes per sample depending on how tough the tissue is.
Cell lysis only takes a few minutes per well, but tissue homogenisation can take 10-20 minutes per sample depending on how tough the tissue is. RNA is stable in trizol which deactivates RNases. You can take a break at this point keeping the sample in trizol for a short time or freezing it for a longer one.
RNA is stable in trizol which deactivates RNases. You can take a break at this point keeping the sample in trizol for a short time or freezing it for a longer one. If supernatant appears turbid an additional chloroform cleaning step can be inserted here.
If supernatant appears turbid an additional chloroform cleaning step can be inserted here.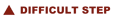 Take care not to aspirate the DNA-containing white interface. This quickly happens and will lead to DNA contamination in your RNA prep.
Take care not to aspirate the DNA-containing white interface. This quickly happens and will lead to DNA contamination in your RNA prep. Do not overdry the pellet or you won’t be able to redissolve it.
Do not overdry the pellet or you won’t be able to redissolve it.
 Human RNA good quality
Human RNA good quality Mouse total RNA undegraded, RIN 10
Mouse total RNA undegraded, RIN 10 Mouse total RNA slightly degraded, RIN 9.4
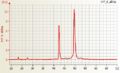
Mouse total RNA slightly degraded, RIN 9.4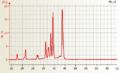 Arabidopsis RNA good quality
Arabidopsis RNA good quality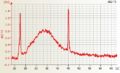 Arabidopsis single cell aRNA
Arabidopsis single cell aRNA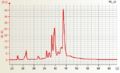 Barley RNA good quality
Barley RNA good quality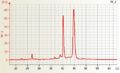 Wheat RNA good quality
Wheat RNA good quality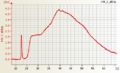 Affymetrix One cycle Human cRNA good quality
Affymetrix One cycle Human cRNA good quality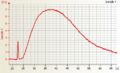 Affymetrix Two cycle Human cRNA good quality
Affymetrix Two cycle Human cRNA good quality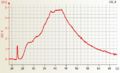 Affymetrix one cycle Arabidopsis cRNA good quality
Affymetrix one cycle Arabidopsis cRNA good quality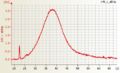 Affymetrix one cycle Arabidopsis single cell aRNA cRNA
Affymetrix one cycle Arabidopsis single cell aRNA cRNA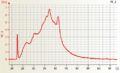 Affymetrix one cycle Barley cRNA good quality
Affymetrix one cycle Barley cRNA good quality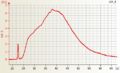 Affymetrix one cycle Wheat cRNA good quality
Affymetrix one cycle Wheat cRNA good quality Affymetrix one cycle Wheat cRNA poor quality
Affymetrix one cycle Wheat cRNA poor quality-PDF")





