Abstract
Nuclear extract preparation is a useful procedure to enrich nuclear proteins for various techniques including EMSA and ChIP studies. There are two separate protocols below;
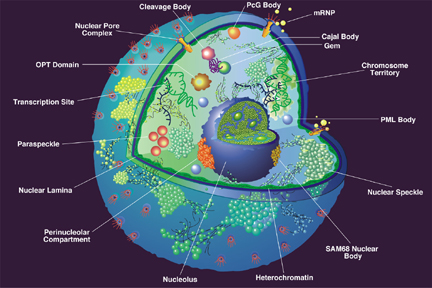 The nucleus of a eukaryotic cell
The nucleus of a eukaryotic cell
Hattori et al nuclear extract preparation
Reagents I
I. Homogenization buffer:
0.3M sucrose 10mM HEPES pH 7.6, 0.1M EDTA 0.1M EGTA 10mM KCl 1mM DTT 0.5mM PMSF in ethanol 0.74mM spermidine 15mM spermine 2mg/ml of each: aprotinin lupeptin bestatin
II. Cushion buffer: (same as homogenization buffer except 2.2M sucrose)
III. Lysis buffer:
10% glycerol 10mM HEPES, pH 7.6 0.1M EDTA 3mM MgCl2 100mM KCl 1mM DTT 0.1mM PMSF in ethanol 0.74mM spermidine 15mM spermine 2mg/ml of each: aprotinin lupeptin bestatin
IV. Dialysis buffer:
20% glycerol 20mM HEPES, pH 7.6 0.2M EDTA 1mM NaMoO4 100mM KCl 2mM DTT 0.1mM PMSF in ethanol 0.74mM spermidine 15mM spermine 2mg/ml of each: aprotinin lupeptin bestatin
4M ammonium sulfate 528g of the salt/l, equal to 75% (saturated) ammonium sulfate
500mM PMSF, 1,000x 870mg of the powder per 10ml of ethanol
Procedure I
I. Homogenize cells or tissue 1. Wash commercially available HeLa cells (30g) or minced in 3mm pieces fresh liver(15-20g) from staining with homogenizing buffer (10ml) and transfer it to 100ml beaker 2. Add to the beaker another 25ml of the buffer and split the content in half (2x15ml) 3. Homogenize each portion twice, 10 strokes (“up” and “down”) each with a teflon pestle. Between strokes, cool homogenizer on ice. Try to maintain temperature close to +4C
II. Pellet the nuclei 1. Filter homogenate through the four- layer cloth (25-30ml) and mix homogenate with cushion buffer (50ml of cushion buffer per 25ml of the homogenate 2. Distribute to ultracentrifuge tubes cushion buffer (10 ml of buffer per a tube). Carefully load cushion buffer- homogenate mix on the top (total volume is about 38 ml). 3 Equilibrate tubes at the balance and centrifuge them in SW28 rotor (24,000 rpm or 76220g, 50min, 1C) or 60Ti rotor (same time and temperature at 32,750 rpm/min)
III. Lyse nuclei 1. Invert tubes and keep them on ice for 10min 2. Resuspend pellets in nuclear lysis buffer (5ml/pellet). Perform one stroke with Dounce homogenizer 3 Add lysis buffer to 40ml and 2ml of 4M ammonium sulfate and agitate for 30-60min at +4C
IV. Reprecipitate nuclear proteins 1. Centrifuge lysates in 55.2Ti rotor (35,000rpm, 60min, +1C) or 60Ti rotor (37,700rpm, 60min, +1C). Save super. 2. Add ammonium sulfate (330mg/ml of solid salt or 65ml of 4M solution) and keep sample(s) on ice for 15min 3 Centrifuge sample(s) in 55.2Ti rotor or 60Ti rotor for 20min at the same speed and temperature. Save THE PELLET(S).
V. Dialyze nuclear extract and measure protein 1. Resuspend each pellet in 500ml of dialysis buffer and dialize sample(s) against dialysis buffer (1:1,000 for 4h) 2 Clarify nuclear extract by centrifugation (+4C, 10min) in microfuge. 3. Measure protein and aliquot supernatant (100-500mg/tube) and store it at -70C
References I
- Hattori M, Tugores A, Veloz L, Karin M, and Brenner DA. A simplified method for the preparation of transcriptionally active liver nuclear extracts. DNA Cell Biol. 1990 Dec;9(10):777-81. DOI:10.1089/dna.1990.9.777 | PubMed ID:2264931 | HubMed [Paper1]
Dignam et al nuclear extract preparation
Reagents II
Buffers used for extract preparation are designated as follows:
Buffer A: 10 mM HEPES (pH 7.9 at 4oC), 1.5 mM MgCl2 , 10 mM KCL and 0.5 mM DTT
Buffer B: 0.3 HEPES (pH 7.9), 1.4 M KCL and 0.03 M MgCL2
Buffer C: 20 mM HEPES (pH 7.9), 25% (v/v) glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride (PMSF) and 0.5 mM DTT
Buffer D: 20 mM HEPES (pH 7.9), 20% (v/v) glycerol, 0.1 M KCL, 0.2 mM EDTA, 0.5 mM PMSF, and 0.5 mM DTT. DTT and PMSF were added fresh to the buffers just before use.
Procedure II
Hela cells were grown in spinner flasks at 37oC in Joklik’s MEM containing 5% calf serum. They were frown to 4 to 6 x 105 cells per ml prior to harvesting for extract preparation.
HeLa cells were harvested from cell culture media by centrifugation (at room temperature) for 10 min at 2000 rpm in a Sorvall HG4L rotor. Pelleted cells were then suspended in five volumes of 4oC phosphate buffered saline and collected by centrifugation as detailed above; subsequent steps were performed at 4oC.
The cells were suspended in five packed cell pellet volumes of buffer A and allowed to stand for 10 min.
The cells were collected by centrifugation as before and suspended in two packed cell pellet volumes (volume prior to the initial wash with buffer A) of buffer A and lysed by 10 strokes of a Kontes all glass Dounce homogenizer (B type pestle).
The homogenate was checked microscopically for cell lysis and centrifuged for 10 min at 2000 rpm in a Sorvall HG4L rotor to pellet nuclei.
The supernatant was carefully decanted, mixed with 0.11 volumes of buffer B, and centrifuged for 60 min at 100,000 gav (Beckman Type 42 rotor). The high speed supernatant from this step was dialyzed five to eight hours against 20 volumes of buffer D and is designated the S100 fraction.
The nuclear extract was prepared as follows. The pellet obtained from the low speed centrifugation of the homogenate was subjected to a second centrifugation for 20 min at 25,000 gav (Sorvall SS34 rotor), to remove residual cytoplasmic material and this pellet was designated as crude nuclei.
These crude nuclei were resuspended in 3 ml of buffer C per 109 cells with a Kontes all glass Dounce homogenizer (10 strokes with a type B pestle). The resulting suspension was stirred gently with a magnetic stirring bar for 30 min and then centrifuged for 30 min at 25,000 gav (Sorvall SS34 rotor).
The resulting clear supernatant was dialyzed against 50 volumes of buffer D for five hours. The dialysate was centrifuged at 25,000 gav (Sorvall SS34 rotor) for 20 min and the resulting precipitate discarded.
The supernatant, designated the nuclear extract, was frozen as aliquots in liquid nitrogen and stored at -80oC.
The protein concentration was usually 6-8 mg/ml and 15-20 mg of protein were obtained from 109 cells.
Reference
- Dignam JD, Lebovitz RM, and Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983 Mar 11;11(5):1475-89. DOI:10.1093/nar/11.5.1475 | PubMed ID:6828386 | HubMed [Paper1]









