 Tubulin loading control Western blot is an important control experiment to know your reagents and protocol are in line and that equal protein amounts are loaded in each lane
Tubulin loading control Western blot is an important control experiment to know your reagents and protocol are in line and that equal protein amounts are loaded in each lane
 No signal on a prolonged exposure could be an issue with either the primary antibody OR protein expression level
No signal on a prolonged exposure could be an issue with either the primary antibody OR protein expression level
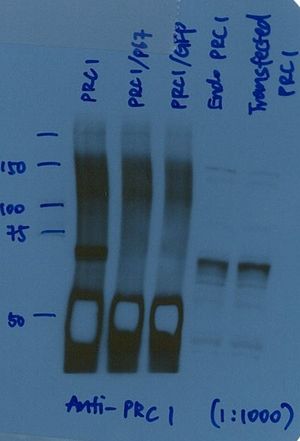 Excessive signal from heavy chain IP/WB @50 kD produces ‘reverse banding’
Excessive signal from heavy chain IP/WB @50 kD produces ‘reverse banding’
Time and labor at the bench is extremely valuable. Below is a guide to interpret western blotting experiences in a purposeful way so that each experiment will make a difference. Watch out for futile cycling in work flow and focus energy in a meaningful way.
There is an increasing number of available commercial antibodies from several vendors. Depending the vendor, the level of quality control is highly variable and this leaves a responsibility to optimize protocols in an efficient manner. Because every antibody is different, each one requires a different blocking/incubation buffer in order to optimize the signal:noise ratio.
There are common themes in the types of incubation buffers that tend to work well for blocking and incubation.
Commercial antibodies have a wide range of specificity and sensitivity for the protein of interest. Efficient blocking of the primary and secondary antibodies can take place in a variety of different blocking agents that contain soluble proteins and non-ionic detergent. When developing a new antibody by western blot, it is important to test different blocking/incubation buffers for the best possible signal:noise ratio in the assay. No single blocking agent is ideal for every primary antibody, since each antibody-antigen pair has unique binding characteristics.
Molecular weight vs. actual protein migration
Western blotting is a technique that separates proteins based on size. In general, the smaller the protein, the faster it migrates through the gel. However, migration is also affected by other factors, so the actual band size observed may differ from that predicted. Common factors include:
1. Post-translational modification – e.g. phosphorylation, glycosylation etc, which increases the size of the protein
2. Post-translation cleavage – e.g. many proteins are synthesized as pro-proteins and then cleaved to give the active form, e.g. pro-caspases
3. Splice variants – alternative splicing may create different sized proteins produced from the same gene
4. Relative charge – the composition of amino acids (charged vs non-charged)
5. Multimers – e.g. dimerization of a protein. This is usually prevented in reducing conditions, although strong interactions can result in the appearance of higher bands
Always run a + control and Secondary control
- Running a + control is helpful in all situations where the banding profile for a given gene may have variation between cell/tissue types. Running the + control will determine if your system protein levels are in question vs. antibody titer/quality.
- Running a secondary control in parallel will ensure that any artifact bands are clearly understood be.
The importance of the blocking/antibody incubation buffer
PVDF or nitrocellulose membrane has the ability to bind protein of all sorts. Since both the antibodies and the transferred lysate/extract are proteins, steps must be taken to optimze interactions between the membrane and the antibody used for detection of the target protein.
Blocking of non-specific binding is achieved by placing the membrane in a dilute solution of protein containing a low percentage of detergent such as Tween-20. The protein in the dilute solution attaches to the membrane in all places where the target proteins have not attached from the gel transfer to the membrane.
When the primary antibody is added, there is no room on the membrane for it to attach other than on the binding sites of the specific target protein. This reduces “noise” in the final product of the Western blot, leading to clearer results.
Band appearance/aesthetics
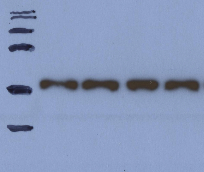 Dumbbell shaped bands are an indication of a hot run
Dumbbell shaped bands are an indication of a hot run
Smile effect of the bands/Dumbbell shaped bands
1. Migration was too fast.
2. Migration was too hot (changing the pH and altering the migration). Slow down the migration or run the gel in the cold room or on ice.
Uneven band size in lanes probed for the same protein
Gel has set too quickly while casting and the acrylamide percentage is not even along the lanes. Review the recipe of the gel and the addition of TEMED to the gels, add a little 0.1% SDS in water to the top of the migrating gel while it sets to stop it from drying.
Molecular weight disparity
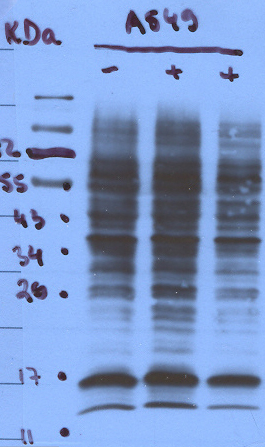 Laddering effects in a western blot may suggest that primary specificity is low and/or the antibody is overly sensitive
Laddering effects in a western blot may suggest that primary specificity is low and/or the antibody is overly sensitive
Always review protein molecular weight details in the primary literature through PubMed for the most informed perspective on your result. Below are possibilities to explain the infinite nuances of protein behavior in SDS-PAGE.
Post-translation modification
- phosphorylation: Tyrosine, Serine, Threonine phosphoryl additions are common.
- glycosylation: Carbohydrate additions due to ER/Golgi processing can increase the weight of the protein.
- cleavage: pro-proteins undergo cleavage to render the active form.
mRNA splice variation
- Alternative splicing of exons can create different sized proteins from the same gene. Depending on cell type, differentiation state, & tissue type, the transcript splicing can and will influence structure and function for certain genes.
Intrinsic structure
- Relative charge: The composition of amino acids (charged vs non-charged).
- Multimers: Modular proteins or proteins than can form multimeric complex can influence the observable weight. This is usually prevented in reducing conditions through the use of DTT, 2-Mercaptoethanol, or TCEP. However, strong interactions can result in the appearance of higher bands.
Black spots on film
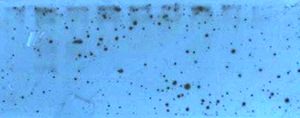 Spotted film below the 140kDa Collagen Type I (COL1A1) precursor band
Spotted film below the 140kDa Collagen Type I (COL1A1) precursor band
- Primary and/or secondary antibody aggregation in solution will cause the film exposure to appear speckled; either primary or secondary antibody. Antibodies may be sticking to the blocking agent.
Microfuge precipitate
- First approach would be to microfuge the primary and secondary at 14,000xG for 15 minutes at 4C.
- 0.2 micron filter sterilize the antibodies into a fresh sterile tube and relabel the tube.
- 0.45 uM filter the blocking agent.
- Proposing to do 14,000xG for 15 minutes at 4C then 0.2um filter supernatant back into the vial.
Remove air bubbles
- use a pasteur pipette as a roller. roll the membrane and gel in the submersed transfer buffer before transfer to remove bubbles. keep membrane and materials wet.
- keep film clean before adding to the developer
Concentration of antigen
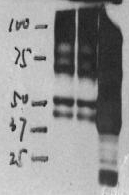 Over abundance of antigen and/or too much primary antibody
Over abundance of antigen and/or too much primary antibody
The resolution of SDS-PAGE is limited to 50-100 bands. If the relative concentration of the antigen of interest is too low (less than 0.2% of total protein), it may be difficult to detect (for instance, synaptobrevin/VAMP comigrates with histones in cell homogenates which interfere with its detection). Signal enhancement may then lead to the appearance of artificial bands. Enrichment of the antigen by fractionation or by immunoprecipitation should be considered.
On the other hand, too much abundance of antigen or too much primary antibody may yield an overly robust signal.
Excess signal; multiple bands/too much banding
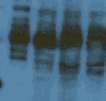 Extraneous banding may represent glycosylation or phosphorylation of the major band
Extraneous banding may represent glycosylation or phosphorylation of the major band
Theoretical prediction is a primary antibody toward a single gene product should produce a single band. While this is common and in some cases to be expected, there are legitimate exceptions to the rule and other factors may be responsible. For example:
Factors that can produce multiple bands of variable molecular weight
- transcript variation
- multiple start/stop sights
- signal-dependent protein processing/shuttling (ie secretion)
- cell type/differentiation state specific variation
- endoplasmic reticulum and golgi-dependent alternative cofactor additions
Smearing effect for the band of interest
- post-translational modifications including glycosylation, nitrosylation, phosphorylation, methylation, acetylation, ubiquitination,
- ubiquitin-dependent protein degradation
Re-applying ECL reagent
The HRP (horseradish peroxidase) is an enzyme that oxidizes the ECL/luminol (contains HRP substrate + enhancing chemicals (modified phenols)). This oxidation reaction with chemiluminescent substrates produces light. The HRP enzyme can remain active as long as it is kept 4C in 1X TBS buffer, no azide. The moment you expose your blot with the HRP-conjugated antibody to the ECL, the substrate begins to be used up but it doesn’t destroy the HRP enzyme.
If banding signal is excessive:
- Place membrane in 1XTBS overnight at 4C. The following day, simply re run the ECL step.
- Blots can be kept in 1X TBS for a 2-3 days 4C. If the signal is too weak, reprobe with the secondary antibody or protein A-HRP, then add ECL reagent.
Excess signal; dark exposure
 Adjust blocking, 1ary, 2ary diluent all to 5% milk TTBS, and perform 10 shake rinses after the 2ary incubation to resolve overexposure of film
Adjust blocking, 1ary, 2ary diluent all to 5% milk TTBS, and perform 10 shake rinses after the 2ary incubation to resolve overexposure of film
 Adjust blocking, 1ary, 2ary diluent to 5% milk TTBS, titration of the primary, further washing after 2ary incubation, and adjusting exposure time can improve the signal:noise
Adjust blocking, 1ary, 2ary diluent to 5% milk TTBS, titration of the primary, further washing after 2ary incubation, and adjusting exposure time can improve the signal:noise
How do I decrease the background on my blot?
The leading cause of excess background is cross-reactivity between blocking agent and primary or secondary antibody: this will result in an overall membrane staining. The best blocking/incubation buffer for your immunoassay is the one that gives you the most clean bands with minimal background noise.
Concentration of antibody may be too high or incubation time too long. Also, several short washing steps are better than one long one.
Additional considerations;
The primary antibody you purchased may be too sensitive for specific detection of the target protein. Contact the vendor and explain your results. Value your time and notify the vendor of your observations. Common feedback may include;
1. Further dilute out your HRP or AP conjugated secondary antibody.
2. Instead of primary antibody incubation overnight, try 2 hours at room temp.
3. Instead of 2 hours at room temp, try 1 hour are room remp or 1 hour at 37C.
4. Perform more wash steps between incubation steps. Try 5 shake rinses followed by 4 x5min washes in 1X TTBS. Several short washing steps are better than one long one.
5. Load less protein onto the gel;
whole cell lysate or tissue extract: 20-50 micrograms subcellular fractions (ie nuclear or cytosol extracts): 10-30 micrograms purified proteins (ie recombinant or eukaryotic expression): 5-50 nanograms
- Blocking of non-specific binding might be absent or insufficient. Increase the blocking incubation period and consider changing blocking agent. *The primary antibody concentration may be too high. Titrate the antibody to the optimal concentration, incubate for longer but in more dilute antibody (a slow but targeted binding is best).
- Incubation temperature may be too high. Incubate blot at 4°C.
- The secondary antibody may be binding non-specifically or reacting with the blocking reagent. Run a secondary control without primary antibody.
- Cross-reaction between blocking agent and primary or secondary. Add a mild detergent such as Tween20 to the incubation and washing buffer.
- Antibody detects the casein present in the milk. Use BSA as a blocking reagent instead of milk.
- Washing of unbound antibodies may be insufficient. Increase the number of washes.
- Your choice of membrane may give high background. Nitrocellulose membrane is considered to give less background than PVDF.
- The membrane has dried out. Care should be taken to prevent the membrane from drying out during incubation.
Ghost bands (reverse/white banding)
 Ghost bands
Ghost bands
Insufficient blocking
Ghost banding is the negative image of the proteins that have been transferred to the membrane.
Visualize in your mind; A 15 lane gel that has been run out and coommassie stained. Every lane will have a laddering appearance. Now imagine this same laddering band pattern being transferred to a PVDF or nitrocellulose membrane. The transfer of the proteins from the gel onto the surface of the membrane creates a protein fingerprint on the membrane. Blocking the membrane is an attempt to cover all other unbound sights.
Ghost banding occurs when there is residual background noise around where the protein fingerprint is present. This event suggests;
1. There is no detectable level of the target protein in the samples
2. The primary antibody is not recognizing the target protein
Excessive signal generated
Reduce the primary/secondary antibody dilution and lower the protein loading amount; Dilute HRP-conjugate at least 10-fold. High levels of specific antibody and an overabundance of protein can cause intense localized signals (typically a single band). Rapid and complete quenching of the substrate will produce no signal. Since there is no light production after the completion of the reaction, white bands are the result when exposed to film.
No signal
 Actin or GAPDH control Western blot is an important control experiment to know your reagents and protocol are in line
Actin or GAPDH control Western blot is an important control experiment to know your reagents and protocol are in line
- Run an antibody control: parallel Actin or GAPDH positive control western blot that corresponds to the same host species as your experimental primary antibody will conclusively indicate if the issue relates to your protocol on some level OR an issue with the primary antibody/protein expression level. This also validates that your secondary antibody is functional.
- Run a positive control: Running a sample that is known to express your protein of interest will conclusively indicate if the issue relates to the primary antibody.
- Antigen is not recognized by primary antibody & this can occur especially with monoclonal antibodies that were raised against a native protein. In some cases, a non-reducing gel system may need to be used. Otherwise contact the vendor technial service.
1. Reagent omitted or improperly prepared. A simple fix yet this becomes more and more rare with experience. Review the protocol.
2. Protein did not transfer from gel to membrane. Try a Ponceau S stain of the membrane to see if there are bands on the membrane.
3. Specificity of HRP secondary antibody not appropriate for primary antibody.
4. Correct orientation of membrane not maintained throughout procedure.
5. Presence of azide in buffer, inhibiting peroxidase activity. Horseradish peroxidase labeled antibodies should not be used in conjunction with sodium azide. A change in the blocking agent or incubation solution will solve this problem.
6. Detergent is too harsh: SDS, Nonidet P-40, and Triton X-100 disrupt binding between proteins. 0.01-0.05% Tween-20 is the most commonly used and recommended detergent for washing and incubation solutions.
Proteolytic breakdown of the antigen
If additional smear/ladder type banding is of lower apparent molecular mass than the full-length protein, then proteases may be active. The addition of fresh protease inhibitors such as PMSF, pepstatin or leupeptin can resolve this. Proteases can mediate degradation when samples are stored for prolonged time or samples are fractionated from starting cell or tissue preps.
Weak signal/poorly defined signal
 Mulitple blank blots for different antibodies suggests there is a need to load positive controls in order to address sample loading amount and sample quality
Mulitple blank blots for different antibodies suggests there is a need to load positive controls in order to address sample loading amount and sample quality
First and foremost, the primary antibody may have low affinity for target protein. Antibody affinity may also change after denaturation of a cell/tissue sample with SDS.
1. Low antibody concentration. Increase the primary dilution.
2. Incubation time needs to be extended.
3. Insufficient protein loaded onto gel. Load more protein.
5. Exposure of film too brief. Try multiple exposures extending from 1 minute all the way to overnight.
6. Bald Spots: bubbles between gel and membrane: bubbles create points of high resistance that lead to low transfer efficiency, be sure to remove bubbles completely when putting together the transfer sandwich.
7. Incomplete Transfer.
One of several technical errors can be the source of incomplete transfer
- Proteins not completely eluted out of gel: this often occurs with high molecular weight proteins, especially when using a transfer buffer containing methanol. One way to overcome this phenomenon is by using nitrocellulose, which does not require methanol in the transfer buffer. Adding SDS to the transfer buffer as well as using higher field strengths also improves protein elution.
- Proteins have transferred through membrane: this may occur when working with proteins of very low molecular weight. Optimizing/shortening transfer times and using a double layer of membrane usually enables retention of small proteins.
- Inappropriate transfer buffer used: the most stable and commonly used buffers are Tris-Glycine based and contain methanol.
- Impurities in the transfer buffer: this will lead to a pattern on the membrane that mirrors the holes in the transfer cassette. Fresh buffer should be prepared prior to each transfer process.






















 Complement Pathway
Complement Pathway