Lentivirus, shRNA plasmid, or siRNA?
Choosing between Lentivirus (somewhat stable), shRNA transfer vector (stable/transient), or siRNA (transient). Perform and exhaustive literature search (pubmed) for citations that have reported RNA/DNA delivery methodology to (species) (lineage) cells. Discuss, evaluate and compare the materials and methods.
RNA (siRNA)
- For easy to transfect cell line, RNA/DNA delivery protocol/procedure should be well defined. siRNA is a proven and effective method targeting mRNA as a molar calculable substrate. In studying effects that can be measured with transient mRNA knockdown, siRNA is a reliable and straightforward approach. Empirical moles of duplex/# cells or total volume (molarity) combines with a minimal number of steps and peripheral controls toward achieving results.
DNA (shRNA)
- shRNA transfer vectors are ~7 kb DNA constructs that will be more stable to handle than RNA. However there are considerable nuances in the actual design and cloning of these vectors, in the actual establishment of antibiotic resistance toward generating a stable population, and in controlling for specific silencing/reproducible results.
- shRNA is a large delivery vector producing a small hairpin substrate over cell passages under antibiotic selective pressure (ie puro resistance may not = Pol III shRNA cassette expression).
- shRNA transfer vector alone may work insofar as culturing the cell, liposomal DNA transfection, antibiotic selection, and then collect data. However the lentivirus transfer vector is engineered for packaging into transducible (VSV-G) pseudotyped viral particles.
Lentivirus
- For primary cell and/or differentiation restrictive (neuron), transducible (VSV-G) pseudotyped viral particles for introducing shRNA (hairpin) is compelling. Measure tropism first with a copGFP reporter.
- Establishing finite passage stable phenotype is feasible with lentiviral particles. (VSV-G) pseudotyped viral particles from Santa Cruz Biotechnology Inc. possess broad tropism.
TECHNICAL SERVICE GUIDE: siRNA
Catalog # Lot #
Summary:
1) Optimize the transfection reagent; measure transfection efficiency of the transfection reagent with FITC-siRNA.
2) Measure knockdown in a range of cell densities ( 30-80%) within 24-72 hours
3) Measure knockdown in a range of siRNA concentrations (30-90 nM) within 24-72 hours
Providing suggestions outlined in the notes below is worth considering and may bring success.
Background Info
- What are the experimental results?
- Describe how gene knockdown is measured? qPCR / Western / IF
- How was the RNA reconstituted?
NOTE: siRNA ships lyophilized along with RNase free water with instructions to reconstitute with 330 ul of H2O to make 10 uM solution. Having the correct molarity of the solution is critical.
- Molarity of siRNA vialed: 10 uM ( uM/L )
- Volume after reconstitution: 330 uL
- Mass of 1 mole of siRNA: 13800 g/mol ( 21nt X 660 g/base pair)
- Total mols per vial: 10 um/L X 330 uL = 3.3 nm
- Total grams per vial: 3.3 nm X 13800 g/mol = 45.5 ug
- Solution concentration: 45.5 ug/ 330 uL = 0.138 ug/uL
- Did this same vial or other lot of siRNA work in the past?
NOTE: If the siRNA same cat# has worked in the past, and now does not work, this may suggest RNase contamination. There are ways to determine this by running 1 pmol (17 ng) siRNA in a native 2% agarose gel, however replacing the vial is a straightforward solution.
Transfection Efficiency
- Describe the cell type for this experiment?
- What transfection reagent is used for the siRNA tranfection?
NOTE: Cationic lipid based transfection reagents (ie Lipofectimine, L2000, Transit TKO, Oligofect, Dharmafect, sc-29528) are each one a unique formula. Certain cell types will respond better to certain cationic lipid (positive charge lipophilic) reagents. For this reason, measuring transfection efficiency is necessary.
- How was transfection efficiency measured?
NOTE: The researcher may have an existing transfection reagent that works on their cells in other experiments (ie cDNA). Suggest to try the same reagent and measure transfection efficiency.
- What time point was transfection uptake of FITC-siRNA measured?
NOTE: Measuring transfection efficiency with sc-36869 will validate that liposome-dependent siRNA entry into the cells is taking place efficiently. It is important to measure transfection efficiency 5-7 hours post transfection since this is when the optimum time point where most transfection takes place. Common methods are IF or Flow cytomtetry.
Cell Confluency
- Adherent cell (grows on the surface of the plate): What is the cell confluency at time of transfection?
- Suspension cell (ie leukocytes/lymphocytes, cells are suspended in the media) : How many cell count # used to seed the well?
NOTE: A hemocytometer (cell counter) is common for counting cells for seeding into multiwell plates (6, 12, 24 well); originally designed for performing blood cell counts. Cell density is an important parameter for knockdown. Optimum cell density will vary and typically falls between 30-80%. NOTE: Setting up a 6 or 12 well experiment and trying a range of cell confluencies (30, 50, 70%), will reveal an optimal cell density where knockdown is optimal with minimal cell death. Effective confluence can range from 30-80%.
siRNA Concentration
- What nanomolar concentration(s) of siRNA are tested?
NOTE: Setting up a 6 or 12 well experiment and trying a range of cell confluencies (30, 50, 70%) & a range of siRNA concentrations (30, 60, 90 nM) will reveal an optimal convergence of cell density and concentration of siRNA where knockdown is optimal with minimal cytotoxicity (cell death).
- What time points is RNAi measured?
NOTE: 48 hours post transfection is a relevant singular point. Measuring knockdown for a few time points in the 24-72 hour window may indicate the frame when RNAi is most optimal. Titrating the siRNA concentration (30-90 nM) for the cells will indicate the best amount to see an effect.
Measuring Knockdown
- How is RNAi measured? Western blot – IF – qPCR – other
NOTE: For WB, titrating the antibody may reveal subtle changes in knockdown. For IF, running secondary controls may indicate nonspecific fluorescence mistaken for signal.
- Quantitative RT-PCR, which primers were used and what type of system?
NOTE: With appropriate internal controls (GAPDH, DNA contamination control), qPCR can be very reliable in determining translation initiation arrest.
TECHNICAL SERVICE GUIDE: Lentivirus
Catalog # Lot #
Summary:1) Determine if the VSV-G coat protein has tropism toward the target cell; measuring transduction efficiency with sc-108084 2) Measure a range of MOI (5-10+) 3) Measure knockdown within 48-72 hours after puromycin selection
Measure transduction efficiency
- Transduction in what cell type?
- Primary cell or Continuous/immortal cell?
NOTE: Primary cell cultures are first generation cells from a living organism and typically have less than 5 passage lifespan. Lenti is popular for primary cells since they are difficult to transfect. Continuous or immortalized cells have the ability to proliferate indefinitely in culture.
- Is this cell type known to have tropism for VSV-G coat protein?
- How is transduction efficiency measured for tropism to VSV-G?
- If a copGFP expressing Lentivirus was used to measure tropism, at what time point was transduction efficiency of the copGFP Control Lentiviral Particles or other reporter measured?
NOTE: 48 hours post-transduction is the time point where puromycin selection begins. This is also a good time point to evaluate transduction efficiency by measuring copGFP fluorescence inside the cells.
- How was the reporter gene measured? (FCM, IF, other)
Multiplicity of Infection (MOI)
- X = uL of virus
- Y = cell count
MOI = X* (particles/uL) /Y
NOTE: A hemocytometer is common for this step; originally designed for performing blood cell counts, contains a etched grid on a slide, count cells/square in 5-10 squares, then average out the number and extrapolate.
NOTE: HEK293T and other easy to transduce cells (MOI of 5-20), while neuronal cells,SHSY5Y, may require MOI of 10-50 particles/cell.
Puromycin Selection
- How many [ug/ml] puromycin is added at what time point post transduction?
- How was optimum puromycin concentration determined?
NOTE: The minimum antibiotic concentration to use is the lowest concentration that kills 100% of non-transfected cells in 3-5 days from the start of puromycin selection (normal range; 1-10 ug/ml). Add puromycin 48 hours post transduction.
- Western blot, IF or Quantitative RT-PCR?
- Negative controls (scrambled hairpin virus, no virus)?
NOTE: Running a parallel transduction with no virus should yield 100% cytotoxicity upon puromycin addition. Scrambled hairpin virus (sc-108080) transduction is useful to determine if any other aspect of the transduction process influences knockdown, including the presence of a non gene specific hairpin (nonspecific antisense).
Transient DNA (shRNA) Transfection
Instead of chemically synthesizing the siRNAs before introducing it in the cell, the siRNAs are made directly by the cells through an expression vector that is transiently transfected into a dividng cell. The shRNA transfer vector alone can be transiently introduced into the dividing cell where the shRNA is synthesized by cellular machinery. While transient transfection is advantageous for fast analysis of shRNA mediated effects, stable transfection ensures long-term, reproducible as well as defined shRNA effects.
Stem Cell ESC Transfection
For DNA transfection of primary cells and sensitive cell lines, Effectene Transfection Reagent is a nonliposomal lipid reagent for DNA transfection into a broad range of cell types. Due to its low cytotoxicity, Effectene Transfection Reagent is suitable on primary or other sensitive lineages.
Stable shRNA Transfection
For many disease models, the most desirable cell types such as immune system or primary cells are not amenable to transfection. Viral delivery of RNAi vectors is a powerful alternative to transfection for these cell types as well as for in vivo applications. Stable expression is achieved by integration of the gene of interest into the target cell’s chromosome: Initially the shRNA of interest has to be introduced into the cell, subsequently into the nucleus, and finally it has to be integrated into chromosomal DNA.
Stable expression can be influenced by two factors: The transfection method used and the vector containing the shRNA of interest. The transfection method determines which cell type can be targeted for stable integration through antibiotic selection. While many lipofection reagents transfect DNA up to a certain amount into adherent cell lines, efficient delivery of DNA into difficult-to-transfect suspension cell lines or even primary cells is only possible with viral methods and nucleofection.
Nucleofection
Nucleofection is a non-viral method of introducing DNA molecules into the nucleus of dividing cells, therefore significantly increasing the chances of chromosomal integration of the transgene. The technology is pioneered by Amaxa
Santa Cruz Biotechnology, Inc. does not disclose vector map information for the sh plasmids, including RE. This removes variable of a single cut RE in the empty showing up in a cloned-in sh,
The tech writing for Lonza nucleofection would suggest a linear plasmid has more efficient outcome for GFP expression , however
- Values are so close between linear and circular DNA for the 2 and 5ug event looking at GFP expression and there is no claim that linear is necessary.
- n= 2 cell lines tested provides limited insight into the validity of the claim for such a device as this : http://www.lonzabio.com/technology.html
- Transgene study only – there is no mention of RNAi or lenti/sh vector suitability to this tech in their nucleofection literature –
Vector dependent
Although there is still some debate as to the effectiveness of this approach, a regular shRNA transfer vector may be able to integrate into the genome of the target cell by antibiotic selection alone. The process may occur randomly by the cell’s machinery itself, possibly via DNA repair and recombination enzymes. If this phenomenon does occur, integration into inactive heterochromatin may result in little or no shRNA expression, whereas integration into active euchromatin may allow for shRNA expression. However, random integration could also lead to silencing of the shRNA cassette. Several strategies have been developed to overcome the negative position effects of random integration: Site-specific, homologous and transposon-mediated integration strategies are used but require the expression of integration enzymes or additional sequences on the plasmid.
Lentiviral particle dependent
Lentiviral particles are highly efficient at infection and stable integration of the shRNA into a cell system. To obtain the lentiviral particle, the transfer vector that contains the shRNA cassette is already flanked by LTRs and the Psi-sequence of HIV. The LTRs are necessary to integrate the shRNA cassette into the genome of the target cell, just as the LTRs in HIV integrate the dsDNA copy of the virus into its host chromosome. The Psi-sequence acts as a signal sequence and is necessary for packaging RNA with the shRNA into pseudovirus particles. Viral proteins which make virus shells are provided in the packaging cell line (HEK 293T), but are not in context of the LTRs and Psi-sequences and so are not packaged into virions. Thus, virus particles are produced that are replication deficient. Lentiviral particles can infect both dividing and nondividing cells because their preintegration complex (virus “shell”) can get through the intact membrane of the nucleus of the target cell.
- Lentiviral systems efficiently transduce both dividing and non-dividing cells
- Study long-term gene knockdown with stable expression
- Reproducibly transduce cell populations
- Inducible or constitutive gene knockdown
Transfection Reagents
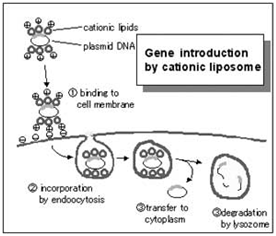
Cationic lipid transfection reagents are suitable for transfecting into a wide variety of dividing cell cultures. Commercial examples include: Lipofectamine / L2000, Dharmafect, iFect, and TransIT TKO. Cationic lipids work by forming lipsomal vesicles that house the siRNA payload and bleb their way through the living cell membrane and into the cytoplasm. The efficiency of this process must be determined in order to have confidence in the knockdown effects. There are numerous commercial sources for transfection reagents for good reason; there are numerous cell types and lipsome structure will influence transfection efficiency in the multitude of experimental cell types that exist.
Polymeric formulations have been developed and optimized for transfection of shRNA plasmid DNA into the nucleus of cultured eukaryotic cells by vendors such as Open Biosystems. Cationic lipids but not polyethylenimine or polylysine prevent transgene expression when complexes are injected in the nucleus (Pollard et al 1998). Polymers but not cationic lipids promote gene delivery from the cytoplasm to the nucleus and transgene expression in the nucleus is prevented by complexation with cationic lipids but not with cationic polymers.
shRNA Vector Transfection
- In a six well tissue culture plate, grow cells to a 50-70% confluency in antibiotic-free normal growth medium supplemented with FBS.
NOTE: This protocol is recommended for a well from a 6 well tissue culture plate. Adjust cell and reagent amounts proportionately for wells or dishes of different sizes.
NOTE: Healthy and subconfluent cells are required for successful transfection experiments. It is recommended to ensure cell viability one day prior to transfection.
Prepare the following solutions:
NOTE: The optimal shRNA Plasmid DNA:shRNA Plasmid Transfection Reagent ratio should be determined experimentally beginning with 1 μg of shRNA Plasmid DNA and between 1.0 and 6.0 μl of shRNA Plasmid Transfection Reagent as outlined below. Once the optimal shRNA Plasmid DNA:shRNA Plasmid Transfection Reagent ratio has been identified for a given cell type, the appropriate amount of shRNA Plasmid DNA/shRNA Plasmid Transfection Reagent complex used per well should be tested to determine which amount provides the highest level of transfection efficiency. For example, if the optimal shRNA Plasmid DNA:shRNA Plasmid Transfection Reagent ratio is 1 μg:1 μl, then amounts ranging from 0.5 μg/0.5 μl to 2.0 μg/2.0 μl should be tested.
Solution A: For each transfection, dilute 10 μl of resuspended shRNA Plasmid DNA (i.e. 1 μg shRNA Plasmid DNA) into 90 μl shRNA Plasmid Transfection Medium (serum antibiotic free medium).
Solution B: For each transfection, dilute 1 – 6 μl of shRNA Plasmid Transfection Reagent with enough shRNA Plasmid Transfection Medium to bring final volume to 100 μl.
NOTE: Do not add antibiotics to the shRNA Plasmid Transfection Medium.
NOTE: Optimal results may be achieved by using siliconized microcentrifuge tubes.
NOTE: Although highly efficient in a variety of cell lines, not all shRNA Plasmid Transfection Reagents may be suitable for use with all cell lines.
- Add the shRNA Plasmid DNA solution (Solution A) directly to the dilute shRNA Plasmid Transfection Reagent (Solution B) using a pipette. Mix gently by pipetting the solution up and down and incubate the mixture 15-45 minutes at room temperature.
- Wash the cells twice with 2 ml of shRNA Transfection Medium. Aspirate the medium and proceed immediately to the next step. NOTE: Do not use PBS as the residual phosphate may compete with DNA and bind the shRNA Plasmid Transfection Reagent, thereby reducing the transfection efficiency.
NOTE: Do not use PBS as the residual phosphate may compete with DNA and bind the shRNA Plasmid Transfection Reagent, thereby reducing the transfection efficiency. For each transfection, add 0.8 ml shRNA Plasmid Transfection Medium to well.
- For each transfection, add 0.8 ml shRNA Plasmid Transfection Medium to well.
- Add the 200 μl shRNA Plasmid DNA/shRNA Plasmid Transfection Reagent Complex (Solution A + Solution B) dropwise to well, covering the entire layer.
- Gently mix by swirling the plate to ensure that the entire cell layer is immersed in solution.
- Incubate the cells 5-7 hours at 37° C in a CO2 incubator or under conditions normally used to culture the cells.
NOTE: Longer transfection times may be desirable depending on the cell line.
- Following incubation, add 1 ml of normal growth medium containing 2 times the normal serum and antibiotics concentration (2x normal growth medium).
- Incubate the cells for an additional 18-24 hours under conditions normally used to culture the cells.
Aspirate the medium and replace with fresh 1x normal growth medium.
- Assay the cells using the appropriate protocol 24-72 hours after the addition of fresh medium in the step above.
NOTE: Controls should always be included in shRNA experiments. Control shRNAs are available as 20 μg. Each encode a scrambled shRNA sequence that will not lead to the specific degradation of any known cellular mRNA.
NOTE: For Western blot analysis prepare cell lysate as follows: Wash cells once with PBS. Lyse cells in 300 μl 1x Electrophoresis Sample Buffer (sc-24945) by gently rocking the 6 well plate or by pipetting up and down. Sonicate the lysate on ice if necessary.
NOTE: For RT-PCR analysis isolate RNA using the method described by P. Chomczynski and N. Sacchi (1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162: 156-159) or a commercially available RNA isolation kit.
Lentivrus Infection
Materials:
- Target cells are SH-SY5Y human neuroblastoma cells.
- Complete medium: DMEM F-12 medium with 10% serum and Penicillin/Streptomycicn
- Vectors are copGFP control Lentiviral Particles (sc-108084)
- Use 24 well plate. Each well has 1.9 mm2, good for working with 0.5 ml medium.
Concentration Considerations:
- Test 5μg/ml Polybrene
- Each well contains 4×104 cells. Will use Viral Particles at a quantity of 4×105 infection units. The concentration of provided viral particles was 5000 infections units per μl, so I will try 100 μl. The MOI is 10.
Day 1 Seed Cells
- Plate target cells in 12 well plate 24 hours prior to viral infection. Each well contains 4×104 cells in 0.5 ml complete medium.
Day 2 Lentiviral Infection
- Monitor the seeded cells and make sure that the cells are around 50% confluent.
- Bring Polybrene (sc-134220) to room temperature, and complete medium to 37°C.
- Prepare a mixture of complete medium with Polybrene at a final concentration of 5 μg/ml.
- Remove media from plate wells and replace with 1ml of Polybrene/media mixture per well.
- Put the plate back to the incubator until lentiviral particles were thawed.
- Thaw lentiviral particles at room temperature and mix gently before use. This takes about 5 minutes.
- Infect cells by adding the indicated amount of shRNA lentiviral particles to the culture.
- Swirl the plate gently to mix and incubate overnight.
NOTE: Lentiviral particles were thawed at room temperature. As soon as the vial is thawed, Lentiviral particles were immediately added to the plate to avoid prolonged exposure of the particles to ambient temperature. Did not use ice.
Day 3 Change Medium
- Remove the culture medium and replace with 1 ml of complete medium (without Polybrene).
- Incubate the cells for 2 days.
- Examine GFP positive cells under microscope. Found that around 80% cells were GFP-positive, the signal was weak.
Day 5 and on Culturing of Cells before Selection
- Starting from Day 5, change to fresh medium and wait for cells to reach confluency in order to get enough cells for the selection.
Changed medium on day5, day8.
shRNA Controls
Negative Controls
- Untreated Cells. Untreated cells will provide a reference point for comparing all other samples.
- Empty construct, containing no shRNA insert; The empty viral particles or DNA are a useful negative control that will not activate the RNAi pathway because it does not contain an shRNA insert. It will allow for observation of cellular effects of the transduction/transfection process. Cells transduced/transfected with the empty control provide a useful reference point for comparing specific knockdown.
- Non-targeting shRNA; This non-targeting shRNA is a useful negative control that will activate RISC and the RNAi pathway, but does not target any human or mouse genes. The short hairpin sequence cotnains 5 base pair mismatches to any known human or mouse gene. This allows for examination of the effects of shRNA transduction/transfection on gene expression. Cells transduced/transfected with the non-target shRNA will also provide useful reference for interpretation of knockdown.
Positive Controls
- Positive shRNA knockdown control; This control contains shRNA sequence that targets GFP expression. This shRNA control has been experimentally shown to reduce GFP expression. This control serves to quickly visualize knockdown in cells expressing GFP.
- Positive shRNA knockdown control; This control contains shRNA sequence that targets eGFP expression (GenBank Accession # pEGFP U476561). The shRNA has been experimentally shown to reduce eGFP expression by 90% in C166-GFP mouse fibroblast cells 48 hours post-transduction by mRNA transcript level. This control serves to quickly visualize knockdown in cells expressing eGFP.
- Positive reporter vector or lentiviral particles; This is a useful positive control for measuring transduction/transfection efficiency and optimizing shRNA delivery. The GFP Control contains a gene encoding GFP, driven by the CMV promoter. This control provides fast visual confirmation of successful transduction/transfection.
copGFP
The copGFP protein is a novel natural green monomeric green fluorescent protein cloned from copepod Pontellina plumata, a type of plankton. The copGFP protein is a non-toxic, non-aggregating protein with fast protein maturation, high stability at a wide range of pH (pH 4-12), and fluorescent properties that do not require any additional cofactors or substrates.
Due to its exceptional properties, copGFP is an excellent fluorescent marker that can be used instead of EGFP (the widely used Aequrea victoria GFP mutant) for monitoring delivery of lentiviral constructs into cells. The copGFP protein has a very bright fluorescence that exceeds the brightness of EGFP by approximately a third.
The copGFP protein emits green fluorescence with the following characteristics:
- emmision wavelength max – 502 nm
- excitation wavelength max – 482 nm
- quantum yield – 0.6
- extinction coefficient – 70,000 M-1 cm-1
When assaying cells, DO NOT fix with methanol and minimize exposure to light. PFA/Formalin fixation works.
Evrogen
Factors Influencing Successful Transfection
Concentration and purity of nucleic acids
Determine the concentration of your DNA using 260 nm absorbance. Avoid cytotoxic effects by using pure preparations of nucleic acids.
DNA:In terms of plasmid preparation, McManus Lab has not observed a need to use E. coli cells that are highly defective for recombination. High DNA quality usually means high transfection efficiency. All DNA preparations should be performed by Cesium prep or endotoxin-free ion exchange plasmid purification methods. If poor transfection is consistently observed, it may be worth performing a additional clean-up of the DNA. The transfection protocols described here are sensitive to the amount of DNA. It is important to optimize DNA:Transfection Reagent ratios.
Transfection in serum-free media
The highest transfection efficiencies can be obtained if the cells are exposed to the transfection complexes in serum free conditions followed by the addition of medium containing twice the amount of normal serum to the complex medium 3–5 hrs post transfection (leaving the complexes on the cells). However, the transfection medium can be replaced with normal growth medium if high toxicity is observed.
No antibiotics in transfection medium
The presence of antibiotics can adversely affect the transfection efficiency and lead to increased toxicity levels in some cell types. It is recommended that these additives be initially excluded until optimized conditions are achieved, then these components can be added, and the cells can be monitored for any changes in the transfection results.
High protein expression levels
Some proteins when expressed at high levels can by cytotoxic; this effect can also be cell line specific.
Cell history, density, and passage number
It is very important to use healthy cells that are regularly passaged and in growth phase. The highest transfection efficiencies are achieved if cells are plated the day before. However, adequate time should be allowed to allow the cells to recover from the passaging (generally >12 hours). Plate cells at a consistent density to minimize experimental variation. If transfection efficiencies are low or reduction occurs over time, thawing a new batch of cells or using cells with a lower passage number may improve the results.
References
- Murphy S, Altruda F, Ullu E, Tripodi M, Silengo L, and Melli M. DNA sequences complementary to human 7 SK RNA show structural similarities to the short mobile elements of the mammalian genome. J Mol Biol. 1984 Aug 25;177(4):575-90. DOI:10.1016/0022-2836(84)90038-x | PubMed ID:6548262 | HubMed [Paper1]
- Czauderna F, Santel A, Hinz M, Fechtner M, Durieux B, Fisch G, Leenders F, Arnold W, Giese K, Klippel A, and Kaufmann J. Inducible shRNA expression for application in a prostate cancer mouse model. Nucleic Acids Res. 2003 Nov 1;31(21):e127. DOI:10.1093/nar/gng127 | PubMed ID:14576327 | HubMed [Paper2]
- Koper-Emde D, Herrmann L, Sandrock B, and Benecke BJ. RNA interference by small hairpin RNAs synthesised under control of the human 7S K RNA promoter. Biol Chem. 2004 Sep;385(9):791-4. DOI:10.1515/BC.2004.103 | PubMed ID:15493873 | HubMed [Paper3]
- Whither RNAi?. Nat Cell Biol. 2003 Jun;5(6):489-90. DOI:10.1038/ncb0603-490 | PubMed ID:12776118 | HubMed [Paper4]
- Pollard H, Remy JS, Loussouarn G, Demolombe S, Behr JP, and Escande D. Polyethylenimine but not cationic lipids promotes transgene delivery to the nucleus in mammalian cells. J Biol Chem. 1998 Mar 27;273(13):7507-11. DOI:10.1074/jbc.273.13.7507 | PubMed ID:9516451 | HubMed [Paper5]



