
{kind=link}
Overview
This is an easy method for screening a large amount of transformants in a cloning procedure, relieving you of the need to worry about optimizing your ligation or doing a huge amount of minipreps. Works best if a vector-only control ligation is transformed as well.
Materials
- Maniatis Sample Buffer III (6x)
- 0.25% Bromophenol Blue
- 0.25% Xylene Cyanol FF
- 30% Glycerol
- Bring up to 100% in H2O
- P2 Lysis Buffer
- 8g NaOH pellets
- 10g SDS
- Bring up to 1L with H2O
- Supercoiled DNA Ladder
Procedure
- Perform your ligation and transformation as usual, plating overnight on selection. You may want to perform a ligation and transformation with a vector-only negative control for comparison purposes.
- The next day, pick single colonies from your vector+insert transformation plates with a toothpick onto a second plate with selection. Spread colony with toothpick into a small (5mm x 5mm) patch on the new plate in a grid, so you can keep individual transformants identified and isolated. I usually pick 30 to 50 colonies at this stage. Incubate this plate at 37°C overnight.
- You may want to pick one or two colonies from your vector only control, if you get any, and patch these as well.
- The next day, cast a large 1% agarose gel, with 1 well per colony you wish to screen (plus a few for your supercoiled ladder). DO NOT add ethidium bromide just yet, as we’ll be running supercoiled DNA, and EtBR affects the mobility of supercoiled DNA.
- Fill however many wells of a 96-well microtiter plate (U-bottom works best) with 10 μL H2O
- Scrape up about half of the patch with a toothpick or sterile loop, and resuspend as best you can in the H2O. If the H2O becomes cloudy, you probably have enough cells suspended.
- Once you’ve finished loading up the wells with cells, use a multipipettor to load 20 μL P2 Lysis Buffer into the wells. Mix briefly with the pipet tip by swirling, NOT pipetting up and down.
 Example result
Example result - Let this sit ~5 minutes
- Use the multipipettor again to load 6 μL Maniatis Sample Buffer III to the wells. Be careful about pipetting up and down to mix, as the lysis mix is pretty sticky, and you don’t want to lose your sample.
- Take your gel out and put it on your bench. Don’t load the gel with it sitting in the buffer, your sample will float out. Load the entire contents of the microtiter wells into the comb (usually you can only get about 30 μl of volume out of these, as they’re pretty gooey by this stage). Don’t worry about bubbles or anything like that in the wells. Remember also to load a supercoiled ladder (not a linear ladder).
- Fill the gel running chamber with your electrophoresis buffer, and carefully lower your loaded gel in. Some of your sample will probably float out, this is okay.
- Run the gel until the bromophenol blue front (dark blue) runs about 3/4 of the way down.
- Stain ~15′ in EtBr, visualize on UV.
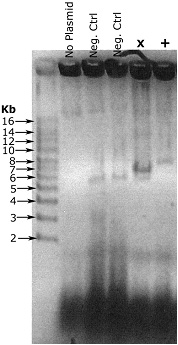
In this example, my vector is 5.5 kb, and the insert is ~1.8 kb. The negative control colonies are recircularized vector, and run at the right size. The lane marked X is a failure. It contains the recircularized vector that we’re not interested in, and while I’m not entirely sure what that strong band is, it’s not what we’re looking for either. The final lane, marked +, is correct, a single band running at around 7.5 kb.
Notes
*Khturner 10:19, 7 December 2006 (EST): The first couple of times you try this, it might be tough to get a feel for the viscosity of the sample at each stage, but once you get the method to work, it pretty much eliminates the need to optimize your ligation and transformation.
Sean Moore Feb. 5, 2007 Is this necessary? This adds a whole day to the screening procedure? You could have done PCR on the colonies in a few hours from the original plate if you needed to screen for colonies with the correct insert. I don’t see more colonies on the “vector + insert” plate than the “vector only” plate, I usually don’t even bother screening and assume something is wrong with the fragments. I have never “optimized” ligations. Also, some replication origins cause significant catenation so analysis in the supercoiled form is impractical/unreliable.
*Khturner 16:51, 15 April 2007 (EDT): You know, you’re probably right. It used to help me when I had problems cloning and a successful construct was rare, but now it’s become pretty routine, and the screening does only take a day by colony PCR. I’ll happily remove this from the protocols page, thanks for the comments.
*Bentley Lim May 9, 2009: Don’t take down this protocol. While many basic cloning experiments use sequenced and up-to-date vectors from companies, etc., many plasmids that still exist have no NCBI entry. Sometimes, it’s impossible to reconstruct the plasmid yourself from papers published before 1990. Therefore, with some cloning experiments, you just need a quick-and-dirty way to see if an insert went in and then you can sequence out to carry out the rest of your cloning procedures.